








专利摘要
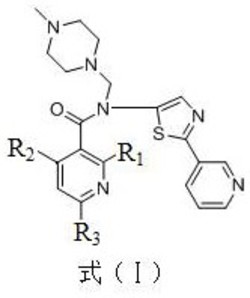
一种新化合物(式1所示)及其盐,实验证明可以抑制肿瘤细胞的生长,用于制备抗肿瘤药物。可以与碱金属Na、K、Me、Ca等生成盐,可以与有机碱甲胺、乙胺、二甲胺、甲醇胺、乙醇胺、氨丁三醇、葡胺、二乙胺、三乙胺等生成盐,具有较好的抑制肝癌和黑色素瘤细胞生长的作用,并经血管刺激性实验证明无溶血及刺激性,可以制备成注射液临床使用。。
权利要求
1.如式(I)所示结构的化合物:
。
2.根据权利要求1所述化合物,与碱金属Na、K、Me、Ca生成的盐,以及与有机碱氨丁三醇、乙醇胺、二乙醇胺、三乙醇胺、二乙胺、三乙胺、葡胺、二甲胺、甲胺等反应得到的盐。
3.权利要求1、2所述化合物在制备治疗肝癌和黑色素瘤药物中的用途。
说明书
技术领域
本发明涉及一种具有抑制肿瘤细胞生长的新化合物,具有较好的水溶性,可以制备成注射液用于治疗肿瘤。
背景技术
肿瘤是危害人类健康的严重疾病,肿瘤的防治工作一直是医药研究领域的重点。目前,由于工业发展中带来的环境污染等问题,人类的生存环境质量不断下降,造成肿瘤疾病的发病率与致死率不断上升。放疗、化疗是目前治疗肿瘤的主要手段。但化疗、放疗在抑制了癌细胞发育的同时也抑制了正常细胞的发育,降低了机体免疫力,导致新的并发症。治疗肿瘤疾病的特效药并不能令人满意,目前临床所用细胞毒性药物选择性不高,导致对正常细胞的恶性杀伤,限制了其应用。因此,寻找新的、无创伤、无细胞毒作用的抗肿瘤药物成为国际医药领域的重要方向。
本发明涉及一种新化合物,经过体外、体内抗肿瘤实验证明具有较好的活性,经过溶解性试验证明具有较好的水溶性,经过溶血和血管刺激性试验证明可以制备成注射液治疗肿瘤。
发明公开
本发明提供了一种抗肿瘤化合物,是一种新的化合物及其盐,具体为式(I)化合物或其药学上可接受的盐:
以及上述化合物碱金属及有机碱反应得到盐。
这种化合物可以通过下面的反应路线取得。
上反应路线中的化合物1、7经过抗肿瘤的体外和体内实验证明具有较好的抑制肿瘤细胞生长的作用。
具体实施例:
实施例1:化合物的制备:
1、化合物4的合成:
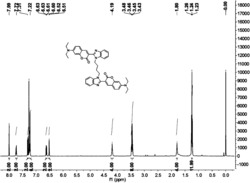
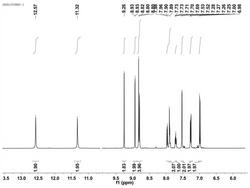
准确称取33.6克化合物3,将其溶于300.0mL的THF和50.0mL的DMF混合溶液中,在冰浴下搅拌30分钟,分批少量的加入4.0克氢化钠(60%),加毕后继续搅拌30分钟。准确称取40.8克化合物2,溶于300.0mL的THF中,将此溶液滴加到上述反应液中,滴加完毕后继续反应1小时,然后自然升温至室温,加热至60℃继续反应3小时。停止反应,加入300.0mL的蒸馏水,然后用饱和氯化铵溶液调节pH至7到8之间。抽滤,滤饼用蒸馏水洗涤(100.0mL×3),乙醇重结晶后得到46.7克化合物4,产率为72%。HNMR(400Hz,DMSO): 8.21(s,1H),8.08(s,1H),7.51(s,1H),7.48(d,J=8.2Hz,2H),7.21(s,1H),7.17(d,J=8.0Hz,1H),7.07(d,J=8.0Hz,1H),6.67(d,J=8.8Hz,2H),6.57(d,J=8.2Hz,2H),6.48(d,J=8.8Hz,2H),4.52(s,2H),4.42-4.40(m,1H),4.21-3.99(m,2H),3.92-3.90(m,2H),3.62(t,J=3.6Hz,4H),3.58(t,J-3.6Hz,4H);MS(m/z):649.5。
2、化合物5的合成:
准确称取32.4克化合物4溶于200.0mL无水乙醇中,在搅拌下加入2.8克氢氧化钾,然后加热至80℃反应30分钟。加入11.3克3-溴丁酮,继续反应5小时。停止反应,冷却反应液至室温,减压蒸馏除去一半体积的溶剂,然后向反应液倒入500.0mL蒸馏水,有沉淀析出,抽滤,滤饼用蒸馏水洗涤,乙酸乙酯重结晶后得到24.3克化合物5,产率为67.5%。HNMR(400Hz,DMSO):8.21(s,1H),8.08(s,1H),7.51(s,1H),7.48(d,J=8.2Hz,2H),7.21(s,1H),7.17(d,J=8.0Hz,1H),7.07(d,J=8.0Hz,1H),6.67(d,J=8.8Hz,2H),6.57(d,J=8.2Hz,2H),6.48(d,J=8.8Hz,2H),4.62(m,1H),4.52(s,2H),4.42-4.40(m,1H),4.21-4.39(m,2H),3.92-3.90(m,2H),3.62(t,J=3.6Hz,4H),3.58(t,J=3.6Hz,4H),2.09(s,3H),1.40(d,J=2.4Hz,3H);MS(m/z):720.5。
3、化合物6的合成:
将21.6克化合物5溶于200.0mL的无水甲醇中,在冰浴中搅拌30分钟,分批少量的加入1.7克硼氢化钠,然后将反应液移至室温下继续反应1小时。停止反应,向反应液加入10.0mL氯化铵饱和溶液和100.0mL蒸馏水,减压蒸馏除去其中的甲醇,再加入100.0mL的蒸馏水,乙酸乙酯萃取(100.0mL L×3),合并有机相,减压蒸馏除 去乙酸乙酯得到17.2可化合物6,产率79.6%。HNMR(400Hz,DMSO):8.21(s,1H),8.08(s,1H),7.51(s,1H),7.48(d,J=8.2Hz,2H),7.21(s,1H),7.17(d,J=8.0Hz,1H),7.07(d,J=8.0Hz,1H),6.67(d,J=8.8Hz,2H),6.57(d,J=8.2Hz,2H),6.48(d,J=8.8Hz,2H),4.52(s,2H),4.42-4.40(m,1H),4.21-4.39(m,2H),4.01(m,1H),3.92-3.90(m,2H),3.84(m,1H),3.62(t,J=3.6Hz,4H),3.58(t,J=3.6Hz,4H),1.30(d,J=2.4Hz,3H),1.21(d,J=2.8Hz,3H);MS(m/z):722.6。
4、化合物7的合成:
将14.4克化合物6,2.5克丁二酸酐,0.06克的DMAP和3.0克三乙胺加入到100.0mL的THF中,在60℃下反应过夜。停止反应,冷却反应液至室温,减压蒸馏除去溶剂得到粗产品,加入100.0mL乙酸乙酯溶解,加入1.0N的稀盐酸30mL,搅拌10分钟后静置分层,有机层用水洗涤(50.0mL×3),无水硫酸钠干燥,减压蒸馏除去溶剂,用乙酸乙酯重结晶后得到15.9克化合物7,产率为98%。90.9%。HNMR(400Hz,DMSO):8.21(s,1H),8.08(s,1H),7.51(s,1H),7.48(d,J=8.2Hz,2H),7.21(s,1H),7.17(d,J=8.0Hz,1H),7.07(d,J=8.0Hz,1H),6.67(d,J=8.8Hz,2H),6.57(d,J=8.2Hz,2H),6.48(d,J=8.8Hz,2H),4.75(m,1H),4.52(s,2H),4.42-4.40(m,1H),4.35(m,1H),4.21-4.39(m,2H),3.92-3.90(m,2H),3.62(t,J=3.6Hz,4H),3.58(t,J=3.6Hz,4H),2.69(t,J=3.2Hz,2H),2.52(t,J=3.2Hz,2H),1.40(d,J=2.8Hz,3H),1.30(d,J=2.4Hz,3H);MS(m/z):822.6。
5、化合物1的合成:
将15.0克的化合物7加入到50.0mL无水乙醇中,在室温、剧烈 搅拌下完全溶解。将1.7克无水碳酸钠溶于10.0mL的蒸馏水中,然后滴加到上述反应液中,这时,反应液中有气泡冒出,待滴加完毕后继续反应1小时。停止反应,减压蒸馏除去一半体积的溶剂,在室温下、搅拌下向反应液滴加100mL丙酮,搅拌30分钟,有沉淀生成。抽滤,滤饼用丙酮洗涤(50.0mL×3),真空干燥后得到12.5克化合物1,产率为81.1%,HPLC纯度为99.0%以上。HNMR(400Hz,DMSO):8.21(s,1H),8.08(s,1H),7.51(s,1H),7.48(d,J=8.2Hz,2H),7.21(s,1H),7.17(d,J=8.0Hz,1H),7.07(d,J=8.0Hz,1H),6.67(d,J=8.8Hz,2H),6.57(d,J=8.2Hz,2H),6.48(d,J=8.8Hz,2H),4.75(m,1H),4.52(s,2H),4.42-4.40(m,1H),4.35(m,1H),4.21-3.99(m,2H),3.92-3.90(m,2H),3.62(t,J=3.6Hz,4H),3.58(t,J=3.6Hz,4H),2.69(t,J=3.2Hz,2H),2.52(t,J=3.2Hz,2H),1.49(d,J=2.8Hz,3H),1.30(d,J=2.4Hz,3H);MS(m/z):844.6。
实施例2:化合物1的抗肿瘤活性
(1)体外抑瘤试验(试验中以代号SZY1402代替实施例1中的化合物1)
实验材料:
药物及试剂:SZY1402
RPMI1640培养基:GIBCO公司生产。
新生牛血清(超级),杭州四季青生物工程材料有限公司。
胰蛋白酶,Sigma公司生产。
细胞株:人肝癌细胞株SMMC-7721、人肺腺癌细胞株A549、人脑胶质瘤细胞株U251、人胃癌细胞株BGC-823
仪器:CO2培养箱(Forma3110,USA),超净工作台(哈东联 BCN-1360B),酶标仪(Bio-Rad550,USA),倒置显微镜(Nikon TE2000,Japan),细胞培养瓶(Costar,USA),96孔细胞培养板(Costar,USA)。
实验方法:
药物及试剂配制:RPMI1640培养基一袋加水一升,补加2克碳酸氢钠,10万单位青霉素和100mg链霉素,调节pH值至7.4,用0.22μm除菌滤膜过滤除菌。95ml培养基加灭活新生牛血清5ml即为完全培养液。胰蛋白酶用D-hanks缓冲液配成0.25%溶液,过滤除菌后4℃保存备用。
准确称取SZY1402 100mg,加到灭菌的1.5ml离心管中,加入DMSO1ml,配成100mg/ml原液,-20℃冷冻保存。临用前融化后取适量以完全培养液稀释成相应浓度应用。
细胞培养及传代:所有细胞均贴壁培养于含10ml完全培养液细胞培养瓶中,于37℃、5%CO2、饱合湿度下培养。细胞长满瓶底后用灭菌D-hanks液洗两次,加0.25%胰蛋白酶消化细胞2分钟,倒掉胰蛋白酶,待轻摇细胞能完全脱落后,加完全培养液30ml后,用移液管吹散细胞,分装于3个新的细胞培养瓶中,继续培养。
药物处理:取刚刚长满的细胞一瓶,胰蛋白酶消化后收集细胞,用移液管吹打均匀,取两滴细胞悬液台盼蓝染色,于显微镜下计数活细胞数目,用完全培养液调整细胞数目至1×105个细胞/毫升。于96孔细胞培养板中每孔加入100μl细胞悬液,将培养板置于CO2培养箱中培养12小时,取出培养板后于每孔中补加100μl含不同浓度SZY1402的完全培养液,使得药物终浓度分别为80.0、60.0、45.0、33.8和25.3μg/ml,每个浓度设4个平行孔,另设4孔细胞加入不含药完全培养液作阴性对照孔,4孔细胞加入含长春新碱的完全培养液 作阳性对照,长春新碱终浓度为5μg/ml。加完药后培养板于微孔板振荡器上振荡混匀,置于CO2培养箱中继续培养48小时。取出培养板,每孔加入10μl 5mg/ml的MTT液,振荡混匀,继续培养4小时,弃去原培养液,于每孔加入DMSO 150μl,充分振荡以溶解蓝紫色结晶,于Bio-Rad 550酶标仪上测定各孔的光吸收,测定波长570nm,参考波长630nm。
根据各孔OD值计算药物对细胞增殖的抑制率:
根据药物浓度的对数对应的抑制率作直线回归,得到直线方程,计算抑制率在50%时对应的药物浓度即为SZY1402对肿瘤细胞的半抑制浓度(IC50)。
于上述同样条件下测定每株细胞的IC50,每株细胞实验连续重复三次。
实验结果:
表1SZY1402对不同肿瘤细胞株的半半抑制浓度(IC50,μg/ml)
(2)体内抑瘤试验
实验材料:
药物及试剂:SZY1402(实施例1中的化合物1),白色粉末。
注射用环磷酰胺,江苏恒瑞医药股份有限公司产品。
动物及瘤株:SPF级昆明种小白鼠,体重18~22g,由山东绿叶制药有限公司动物中心提供,合格证号:SYXK(鲁)20030020。SPF级C57BL/6近交系小鼠,体重18~22g。小鼠H22肝癌、B16黑色素瘤均引自中国医学科学院北京药物研究所。
仪器:CO2培养箱(Forma 3110,USA),超净工作台(BCN-1360,哈尔滨东联),倒置显微镜(Nikon)。
实验1:H22肝癌
H22肝癌于昆明种小鼠腹腔接种后取腹水传代保存。取腹水传代第10日的H22肝癌荷瘤小鼠,脱颈椎处死小鼠,消毒腹部皮肤,以无菌注射器吸取乳白色腹水,以注射用生理盐水调整肿瘤细胞浓度为1×107细胞/ml。以酒精棉球消毒昆明种小鼠右侧腋下皮肤,于皮下接种上述瘤细胞悬液0.2ml,常规饲养。
分组和给药:荷瘤小鼠50只,按体重随机分为5组,每组10只,分别为模型组、环磷酰胺组、SZY1402的10、30、90mg/kg组。各组小鼠按表2所示剂量和方式给药,环磷酰胺组于荷瘤第二天仅腹腔给予一次环磷酰胺,SZY1402组均每日尾静脉给药1次,连续10天。给药体积20ml/kg体重。末次给药后24小时,脱颈椎处死小鼠,称体重,剥取瘤组织称重,计算抑瘤率。
结果以 表示,以t检验进行组间统计学差异比较。
结果显示,30mg/kg的SZY1402连续静脉注射10天,对小鼠移植性肿瘤H22肝癌的生长具有抑制作用。见表2:
表2SZY1402对小鼠H22肝癌生长的抑制作用
与模型组比较:*,P<0.05;**,P<0.01。
实验2:B16黑色素瘤
B16黑色素瘤于C57BL/6小鼠腋下皮下接种传代保存。选择肿瘤生长良好、无坏死或液化的腋下传代保存的B16黑色素瘤荷瘤小鼠,脱颈椎处死小鼠,75%酒精浸泡消毒后,无菌取瘤组织,加入5倍体积(W/V)的注射用生理盐水,用组织匀浆器制研磨成匀浆,以灭菌的200目不锈钢筛网过滤得瘤细胞悬液。同上接种于C57BL/6小鼠腋窝皮下,常规饲养。
分组和给药:荷瘤小鼠50只,按体重随机分为5组,每组10只,分别为模型组、环磷酰胺组、SZY1402的10、30、90mg/kg组。各组小鼠按表3所示剂量和方式给药,环磷酰胺组于荷瘤第二天仅腹腔给予一次环磷酰胺,SZY1402的各组均每日尾静脉给药1次,连续10天。给药体积20ml/kg体重。末次给药后24小时,脱颈椎处死小鼠,称体重,剥取瘤组织称重,计算抑瘤率。
结果以 表示,以t检验进行组间统计学差异比较。
结果显示,30mg/kg的SZY1402组连续静脉注射10天,对小鼠移 植性肿瘤B16黑色素瘤的生长具有抑制作用。见表3:
表3 SZY1402对小鼠B16黑色素瘤生长的抑制作用
与模型组比较:*,P<0.05;**,P<0.01。
实施例4:采用实施例1中化合物1制备的注射液:
按处方量准确称取实施例1制备的化合物1置一容器中,加适量注射用水,搅拌至全溶,并调节pH至8.5-8.8,加注射用水至4000ml,加入2g针用活性炭,煮沸15min,抽滤脱碳,溶液经0.22μm微孔滤膜过滤,溶液灌封于玻璃安瓿内(每支含化合物1:94mg)制剂经115℃加压灭菌30min即可。
实施例5:实施例4中注射液溶血性及刺激性试验
液溶血性试验:
体外溶血试验:向盛有2%红血球混悬液的各支药液管中分别加入不等量的实施例4制备的注射液的低浓度和高浓度(0.63mg/mL和1.88mg/mL),各支药液管在3小时内不产生溶血作用。说明实 施例4制备的注射液体外溶血性试验阴性。具体的实验方法和实验结果如下:
1、受试药物的配制:
(1)高剂量组:取实施例4制备的注射液(4mL:94mg/瓶)1瓶,吸出0.5mL后用0.9%(0.9g/100ml)氯化钠注射液稀释至6.25mL,使成浓度为1.88mg/mL的溶液。
(2)低剂量组:取上述浓度为1.88mg/mL的溶液2mL,用0.9%(0.9g/100ml)氯化钠注射液稀释至6mL,稀释成浓度为0.63mg/mL的溶液。
2、给药方法:
(1)2%红血球混悬液的制备:
取兔血数毫升,放入盛有含玻璃珠的三角瓶中振摇10分钟,除去纤维蛋白原,使成脱纤血液。然后分装在数支离心管中,每管加约10倍量的0.9%氯化钠注射液,摇匀,离心(1500转/分,15分钟),除去上清液,沉淀的红血球再用0.9%氯化钠注射液洗涤2-3次,直至上清液不显红色为止。将所得红血球用0.9%氯化钠注射液配成2%的混悬液,供试验用。
取口径大小均匀的洁净试管7只(每管均设平行管),编号后,用移液管按表9所示配比量依次加入2%红血球混悬液、0.9%氯化钠注射液、注射用水和受试药液,混匀后立即置37℃恒温箱中进行温育,开始每隔15分钟观察一次,1小时后,每隔1小时观察一次,共观察3小时。见表5:
表5:2%红血球混悬液的制备编号
注:其中1-5管为供试品管,第6管为阴性对照管,第7管为阳性对照管。
(2)结果观察:
如试验中溶液呈澄明红色,管底无细胞残留或有少量红细胞残留,即表示有溶血发生;如红细胞全部下沉,上清液体无色澄明,表明无溶血发生。如溶液中有棕红色或红棕色絮状沉淀,振摇后不分散,表明有红细胞凝聚发生。如有红细胞凝聚的现象,需进一步判定是真凝聚还是假凝聚。若凝聚物在试管振荡后又能均匀分散,或将聚集物放在载波片上,在盖玻片边缘滴加2滴0.9%氯化钠注射液,在显微镜下观察,凝聚红细胞能被冲散者为假凝聚;若凝聚物不被摇散或在玻片上不被冲散者为真凝聚。
3、结果判定
当阴性对照管无溶血和凝聚发生,阳性对照管有溶血发生时,若受试物管中的溶液在3小时内不产生溶血和凝聚,则受试物可以注射使用;若受试物管中的溶液在3小时内产生溶血和(或)凝聚,则受试物不宜注射使用。
4、试验结果
分别加入低浓度为0.63mg/mL和高浓度为1.88mg/mL的注射液溶液的各支药液管在3小时内均不产生溶血作用,体外溶血性试验阴性。详见下表6和表7。
表6:注射液(高剂量组)溶血性试验结果(肉眼观察)
注:“+”表示全溶血,“-”表示不溶血;第6管为阴性对照管,第7管为阳性对照管。
表7:注射液(低剂量组)溶血性试验结果(肉眼观察)
注:“+”表示全溶血,“-”表示不溶血;第6管为阴性对照管,第7管为阳性对照管。
注射液血管刺激性试验
兔血管刺激性试验:试验选用健康新西兰兔8只,采用同体左右侧耳朵自身对比法,左侧耳缘静脉注射受试药物,给药体积5ml/kg体重,各给药组给予相应剂量的实施例4制备的注射液,低剂量组和高剂量组剂量分别是3.15mg/kg·bw和9.4mg/kg·bw,(按浓度计算其低浓度和高浓度分别为0.63mg/mL和1.88mg/mL,是临床一次静脉滴注拟用浓度的0.7-1.4倍和2-4倍),右耳给予等体积0.9%(0.9g/100ml)氯化钠注射液作对照,每天一次,连续3天。8只兔依次给予受试药的高浓度和低浓度后,再分别给予0.9% 氯化钠注射液。各取低剂量和高剂量的2只兔于末次给药后48小时剖检,余下低浓度和高浓度的4只兔在末次给药2周恢复期结束后剖检。结果8只动物双耳血管轮廓较清晰,兔耳厚薄均匀,未见明显改变;病理组织学检查,动物双耳血管未见有毒理学意义的改变。说明实施例4制备的注射液血管刺激性试验符合规定。具体的实验方法和实验结果如下:
1、受试物的配制:
(1)高剂量组:取实施例4制备的注射液(4mL∶94mg/瓶)2瓶,吸出8mL后用0.9%(0.9g/100ml)氯化钠注射液稀释至100.0mL,使成浓度为1.88mg/mL的溶液。
(2)低剂量组:取上述浓度为1.88mg/mL的溶液30mL,用0.9%氯化钠注射液稀释至90.0mL,稀释成浓度为0.63mg/mL的溶液。
2、动物称重:给药前及末次给药后48小时和14天各称重一次。
3、一般观察与动物取材:
每天给药前观察并记录动物和血管注射部位的反应,末次给药后48小时,分别放血处死受试药物的高浓度和低浓度的2只新西兰兔,肉眼观察并记录血管组织的反应后,从耳根部剪下双兔耳(先剪左耳,后剪右耳,并标记),然后分别剪取一段兔耳标本固定在10%中性甲醛溶液中(标本长约8cm,宽约1cm;远心端切口距第一针眼约0.5cm处,近心端切口距第三针眼约2cm处,挂线端为近心端)。各留下受试药物的高浓度和低浓度2只动物继续观察至末次给药后14天,进行如下病理检查:以第一针眼为界,远端切一段;以第三针眼为界,近端切二段;制片时血管横切,常规石蜡制片,切片厚度约4-5μm,H-E染色,然后进行病理组织学检查。
4、结果判定
根据肉眼观察和病理检查的结果进行综合判断。
5、试验结果
5.1肉眼观察:
每天给药前肉眼观察并记录动物血管注射部位的反应,给药期间肉眼可见受试药物高浓度和低浓度的部分动物给药侧和对照侧兔耳进针部位血管表皮内外侧呈红色,面积由0.1cm×0.2cm至0.2cm×1.0cm。在末次给药后48小时,受试药物的高浓度和低浓度的4只兔的双侧兔耳血管轮廓较清晰,兔耳厚薄均匀,未见明显改变,详见表12和表13。末次给药后14天剖检受试药物的高浓度和低浓度的4只兔,双侧兔耳血管轮廓较清晰,兔耳厚薄均匀,未见明显改变。
5.2病理检查:
受试药物的高浓度和低浓度的4只兔于末次给药后48小时剖检,余下受试药物的高浓度和低浓度的4只兔在2周恢复期结束后剖检。病理组织学检查均未见血管组织有变性或坏死等显著刺激性反应。结果见表8和表9:
表8:注射液(高剂量组)对兔耳血管刺激反应(末次给药后48小时肉眼观察结果)
表9:注射液(低剂量组)对兔耳血管刺激反应(末次给药后48小时肉眼观察结果)
以上结果说明采用实施例1中的化合物1制备的注射液经过溶血性和血管刺激性试验表明具有良好的安全性,适宜制备成注射液在临床使用。
一种三唑类化合物专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0