

IPC分类号 : C07F9/6584,B01J31/02,C07C211/52,C07C209/42






专利摘要
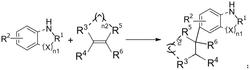
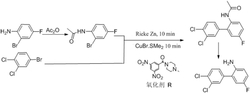
本发明属于有机化工技术领域,特别是一种作为有机催化剂的新型四环稠合磷化物,所述化合物化学结构式为:以邻叠氮苯胺和邻溴硝基苯为原料,在金属催化剂的作用下先后经过碳氮偶联,亚胺化,还原,环化等多个反应步骤得到一种新型四环稠合磷化物。该发明关键点在于合成了一类新颖的四环稠合磷化物;以及制备这类磷化合物的全新合成路线;同时,成功取得了该类四环稠合磷化物有效应用,即能有效的催化Staudinger还原反应。相比于传统的有机磷催化剂,所述的四环稠合磷化物因其四个环的共同作用,致使膦原子的电子较为特殊,使得该四环稠合磷化物在催化有机反应体系中有着显著地作用,能有效的催化Staudinger反应等重要的有机反应。
权利要求
1.一种四环稠合磷化物,其特征在于,所述化合物的化学结构式为:
其中,取代基R
2.合成权利要求1所述的四环稠合磷化物的方法,其特征在于,所述方法包括以下合成路径:
所述方法包括以下步骤:
(1)在反应器中依次加入原料2,溶剂DMSO,K2CO3和邻叠氮苯胺1,将温度升至30-180℃反应6-48h,然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3;
(2)将橙色固体3置于反应瓶内用甲苯溶解,加入三苯基膦,于室温下反应1-24h重结晶得到中间体4;
(3)将含醛或酮的羰基化合物用甲苯溶解,然后与中间体4混合,加热到30-130℃的环境下反应1-48h后,减压除去溶剂得粗中间体5,然后加入乙醇溶液,同时加入硼氢化钠,于室温下反应1-24h,然后将混合物用柱层析纯化,得到中间体6;
(4)将三氯化磷滴加至二氯甲烷中,降温至-60~-80℃,然后将中间体6溶解在二氯甲烷中,用注射器缓慢加至反应液中,再加入吡啶在此温度下搅拌1-24h,移至室温滤出白色沉淀,减压除去溶剂后加入乙醚溶液,在低温下重结晶析出白色晶体7。
3.根据权利要求2所述的方法,其特征在于:所述步骤(1)化合物2与K2CO3和邻硝基苯胺1的投料摩尔比为1:0.1-2:0.1-2;反应温度为120℃;反应时间为36h。
4.根据权利要求2所述的方法,其特征在于:所述步骤(2)化合物3与三苯基膦的投料摩尔比为1:0.1-6;反应时间为12h。
5.根据权利要求2所述的方法,其特征在于:所述步骤(3)化合物4与羰基化合物的投料摩尔比为1:0.1-4;反应温度为110℃;反应时间为24h。
6.根据权利要求2所述的方法,其特征在于:所述步骤(3)中间体5与硼氢化钠的投料摩尔比为1:0.1-4;反应时间为12h。
7.根据权利要求2所述的方法,其特征在于:所述步骤4)化合物6与三氯化磷、吡啶的投料摩尔比为1:0.1-2:0.1-6;反应温度为-78℃;反应时间为12h。
8.权利要求1所述的四环稠合磷化物作为催化剂在催化Staudinger还原反应上的应用。
说明书
技术领域
本发明涉及有机化工技术领域,特别是一类新型四环稠合磷化物、合成方法及其催化应用。
背景技术
有机催化是有机化学中一类非常重要的一个领域,目前已报道的有机催化的反应实例中,催化剂一般都为亚胺和胺等含氮类有机催化剂,而含磷类有机催化剂目前报道的较少。然而,磷原子因其特殊的电子结构赋予了有机磷试剂非常特殊的化学性质,在许多重要的反应中都有广泛的应用,例如:Wittig反应、Staudinger反应、Mitsunobu反应和Appel反应等经典的人名反应都是由有机磷类化合物发起的反应。然而参与这些重要的反应的膦试剂类型较为单一,一般都是三苯基膦、三丁基膦。发展结构新颖,性能优良的新型有机膦催化剂也就显得尤为迫切。在本专利中,我们发展了一类结构新颖的四环稠合磷化物。该四环稠合磷化物因四个环稠合作用,致使活性中心磷原子电子结构较为特殊,从而赋予了该化合物良好的催化活性。同时,合成该四环稠合磷化物的所有制备步骤都是常规的方法,具有较高的可操作性,有利于其实现大规模制备,以及工业化应用。这些特性让这类四环稠合磷化物具有其他磷类催化剂无法比拟的优势,有着良好的应用前景。
发明内容
本发明的主要目的在于提供一类新型四环稠合磷化物、合成方法及其催化应用。
本发明的技术方案如下:
一种四环稠合磷化物,所述化合物的化学结构式为:
其中,取代基R
合成所述的四环稠合磷化物的方法,所述方法包括以下合成路径:
所述方法包括以下步骤:
(1)在反应器中依次加入原料2,溶剂DMSO,K2CO3和邻叠氮苯胺1,将温度升至30-180℃反应6-48h,然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3;
(2)将橙色固体3置于反应瓶内用甲苯溶解,加入三苯基膦,于室温下反应1-24h重结晶得到中间体4;
(3)将含醛或酮的羰基化合物用甲苯溶解,然后与中间体4混合,加热到30-130℃的环境下反应1-48h后,减压除去溶剂得粗中间体5,然后加入乙醇溶液,同时加入硼氢化钠,于室温下反应1-24h,然后将混合物用柱层析纯化,得到中间体6;
(4)将三氯化磷滴加至二氯甲烷中,降温至-60~-80℃,然后将中间体6溶解在二氯甲烷中,用注射器缓慢加至反应液中,再加入吡啶在此温度下搅拌1-24h,移至室温滤出白色沉淀,减压除去溶剂后加入乙醚溶液,在低温下重结晶析出白色晶体7。
进一步优选为:
(1)在反应器中依次加入原料2,溶剂DMSO,K2CO3和邻叠氮苯胺1,将温度升至120℃反应36h。然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3;
(2)将3置于反应瓶内用甲苯溶解,加入三苯基膦,于室温下反应12h重结晶得到中间体4;
(3)将各种醛或酮等羰基化合物用甲苯溶解,然后与中间体4混合,加热到110℃的环境下反应24h后,减压除去溶剂得粗中间体5,然后加入乙醇溶液,同时加入硼氢化钠,于室温下反应6h,然后将混合物用柱层析纯化,得到中间体6;
(4)将三氯化磷滴加至二氯甲烷中,降温至-78℃,然后将中间体6溶解在二氯甲烷中,用注射器缓慢加至反应液中,再加入吡啶在此温度下搅拌12h,移至室温滤出白色沉淀,减压除去溶剂后加入少量乙醚溶液,在低温下重结晶析出白色晶体7。
完成新型四环稠合磷化物的合成。
所述步骤1)化合物2与K2CO3和邻硝基苯胺1的投料摩尔比为1:0.1-2:0.1-2;反应温度为30-180℃,在此反应温度下有利于反应平稳进行,减少副产物生成。反应时间为6-48h,反应时间过长或过短都会对反应的收率有影响。
所述步骤2)化合物3与三苯基膦的投料摩尔比为1:0.1-6;反应时间为1-24h,反应时间过长或过短都会对反应的收率有影响。
所述步骤3)化合物4与羰基化合物的投料摩尔比为1:0.1-4;反应温度为30-130℃,在此反应温度下有利于反应平稳进行,减少副产物生成。反应时间为1-48h,反应时间过长或过短都会对反应的收率有影响。
所述步骤3)中间体5与硼氢化钠的投料摩尔比为1:0.1-4;反应时间为1-48h,反应时间过长或过短都会对反应的收率有影响。
所述步骤4)化合物6与三氯化磷、吡啶的投料摩尔比为1:0.1-2:0.1-6;反应温度为-78-30℃,在此反应温度下有利于反应平稳进行,减少副产物生成。反应时间为1-24h,反应时间过长或过短都会对反应的收率有影响。
本发明有益效果如下:
1、本发明合成一类作为有机催化剂的新型四环稠合磷化物,可有效地替代普通的膦催化剂,作为一种新型有机催化剂广泛运用于众多催化循环的反应体系中,并具有经济环保的优势。
2、本发明设计了一条作为有机催化剂的新型四环稠合磷化物的合成路线。该路线以常见的邻叠氮苯胺和邻叠氮溴苯为原料,先后经过偶联,亚胺化,还原,环化等几个步骤合成最终的新型四环稠合磷化物。该方法产率高,副产物少,有较高的使用价值。
3、本发明所合成一类新型四环稠合磷化物,具有很好的催化活性,在催化Staudinger还原反应时表现出了良好的催化性能。
具体实施方式
下面结合实施例来进一步说明本发明,但本发明要求保护的范围并不局限于实施例表述的范围。
仪器及试剂:
SHZ-E型循环水式真空泵(上海荣亚生化学仪器厂);DZE-6120型真空干燥箱(上海恒天科学仪器制造公司);WRS-1A数字熔点仪(上海索光光电技术有限公司);EB2005A电子天平;ZF-I型三用紫外分析仪;DE-102J集热式恒温加热磁力搅拌器(巩义市华发化学仪器厂);DFX-5L/30低温恒温反应浴(无锡市百川仪器厂);2YZ-4A型旋片式真空油泵(临海市永昊真空设备厂)。频呐酮(AR),甲基异丙基酮(AR),液溴(AR),乙醚(AR),二氯甲烷(AR),三乙胺(AR),苄胺(AR),甲苯(AR),钯碳(AR),甲醇(AR),磷酸钾(AR),三氯化磷(AR),戊烷(AR),去离子水(自制),工业用氮气(AR),工业用氢气(AR)。
实施例1
一种合成5,7-bis(1-(4-chlorophenyl)ethyl)-5,7-dihydrobenzo[d]benzo[4,5][1,3,2]diazaphospholo[1,2-a][1,3,2]diazaphosphole的方法,包括以下实验步骤:
在反应器中依次加入原料邻叠氮溴苯2a(11mmol),溶剂DMSO(50ml),K2CO3(12mmol)和邻叠氮苯胺1a(10mmol),将温度升至120℃反应36h。然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3a(9.3mmol,93%.);将3a(9.3mmol.)置于反应瓶内用甲苯(50ml)溶解,加入三苯基膦(20mmol)于室温下反应12h重结晶得到中间体4a(8.28mmol,89%);将中间体4a(8.28mmol)用甲苯(50ml)溶解之后与对氯苯乙酮(18.26mmol)混合,加热到110℃的环境下反应24h后,减压除去溶剂得粗中间体5a(5.30mmol,64%),然后加入乙醇(50ml)溶液,同时加入硼氢化钠(11.66mmol),于室温下反应6h,然后将混合物用柱层析纯化,得到中间体6a(4.77mmol,90%);将三氯化磷(5.25mmol)滴加至二氯甲烷(30ml)中,降温至-78℃,然后将中间体6a(4.77mmol)溶解在二氯甲烷(20ml)中,用注射器缓慢加至反应液中,再加入吡啶(15.74mmol)在此温度下搅拌12h,移至室温滤出白色沉淀,减压除去溶剂后加入少量乙醚溶液,在低温下重结晶析出白色晶体7a(1.25g,2.48mmol,52%)。
实施例2
一种合成3,9-dichloro-5,7-bis(naphthalen-1-ylmethyl)-5,7-dihydrobenzo[d]benzo[4,5][1,3,2]diazaphospholo[1,2-a][1,3,2]diazaphosphole的方法,包括以下实验步骤:
在反应器中依次加入原料4-氯-2-叠氮溴苯2b(11mmol),溶剂DMSO(50ml),K2CO3(12mmol)和4-氯-2-叠氮苯胺1b(10mmol),将温度升至120℃反应36h。然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3b(9.4mmol,94%.);将3b(9.4mmol.)置于反应瓶内用甲苯(50ml)溶解,加入三苯基膦(20mmol)于室温下反应12h重结晶得到中间体4b(8.46mmol,90%);将中间体4b(8.46mmol)用甲苯(50ml)溶解之后与1-萘甲醛(18.61mmol)混合,加热到110℃的环境下反应24h后,减压除去溶剂得粗中间体5b(6.85mmol,81%),然后加入乙醇(50ml)溶液,同时加入硼氢化钠(15.08mmol),于室温下反应6h,然后将混合物用柱层析纯化,得到中间体6b(5.62mmol,82%);将三氯化磷(6.18mmol)滴加至二氯甲烷(30ml)中,降温至-78℃,然后将中间体6b(5.62mmol)溶解在二氯甲烷(20ml)中,用注射器缓慢加至反应液中,再加入吡啶(18.54mmol)在此温度下搅拌12h,移至室温滤出白色沉淀,减压除去溶剂后加入少量乙醚溶液,在低温下重结晶析出白色晶体7b(1.78g,3.09mmol,55%)。
实施例3
一种合成9-chloro-1-methoxy-5,7-dimethyl-3-(6-methylheptyl)-5,7-dihydrobenzo[d]benzo[4,5][1,3,2]diazaphospholo[1,2-a][1,3,2]diazaphosphole的方法,包括以下实验步骤:
在反应器中依次加入原料4-氯-2-叠氮溴苯2c(11mmol),溶剂DMSO(50ml),K2CO3(12mmol)和6-甲氧基-4-异辛基-2-叠氮苯胺1c(10mmol),将温度升至120℃反应36h。然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3c(6.8mmol,68%.);将3c(6.8mmol.)置于反应瓶内用甲苯(50ml)溶解,加入三苯基膦(15mmol)于室温下反应12h重结晶得到中间体4c(6.26mmol,92%);将中间体4c(6.26mmol)用甲苯(50ml)溶解之后与甲醛(20.64mmol)混合,加热到110℃的环境下反应24h后,减压除去溶剂得粗中间体5c(5.63mmol,90%),然后加入乙醇(50ml)溶液,同时加入硼氢化钠(12.39mmol),于室温下反应6h,然后将混合物用柱层析纯化,得到中间体6c(4.28mmol,76%);将三氯化磷(4.71mmol)滴加至二氯甲烷(30ml)中,降温至-78℃,然后将中间体6c(4.28mmol)溶解在二氯甲烷(20ml)中,用注射器缓慢加至反应液中,再加入吡啶(14.12mmol)在此温度下搅拌12h,移至室温滤出白色沉淀,减压除去溶剂后加入少量乙醚溶液,在低温下重结晶析出白色晶体7c(0.91g,2.10mmol,49%)。
实施例4
一种合成3,9-dichloro-5,7-bis(naphthalen-1-ylmethyl)-5,7-dihydrobenzo[d]benzo[4,5][1,3,2]diazaphospholo[1,2-a][1,3,2]diazaphosphole的方法,包括以下实验步骤:
在反应器中依次加入原料4-氯-2-叠氮溴苯2b(110mmol),溶剂DMSO(500ml),K2CO3(120mmol)和4-氯-2-叠氮苯胺1b(100mmol),将温度升至120℃反应36h。然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3b;将3b置于反应瓶内用甲苯(500ml)溶解,加入三苯基膦(200mmol)于室温下反应12h重结晶得到中间体4b;将中间体4b用甲苯(500ml)溶解之后与1-萘甲醛(186.1mmol)混合,加热到110℃的环境下反应24h后,减压除去溶剂得粗中间体5b,然后加入乙醇(500ml)溶液,同时加入硼氢化钠(150.8mmol),于室温下反应6h,然后将混合物用柱层析纯化,得到中间体6b;将三氯化磷(61.8mmol)滴加至二氯甲烷(300ml)中,降温至-78℃,然后将中间体6b溶解在二氯甲烷(200ml)中,用注射器缓慢加至反应液中,再加入吡啶(185.4mmol)在此温度下搅拌12h,移至室温滤出白色沉淀,减压除去溶剂后加入少量乙醚溶液,在低温下重结晶析出白色晶体7b(39.1%)。
实施例5
一种合成3,9-dichloro-5,7-bis(naphthalen-1-ylmethyl)-5,7-dihydrobenzo[d]benzo[4,5][1,3,2]diazaphospholo[1,2-a][1,3,2]diazaphosphole的方法,包括以下实验步骤:
在反应器中依次加入原料4-氯-2-叠氮溴苯2b(1.1mmol),溶剂DMSO(5ml),K2CO3(1.2mmol)和4-氯-2-叠氮苯胺1b(1mmol),将温度升至120℃反应36h。然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3b;将3b置于反应瓶内用甲苯(5ml)溶解,加入三苯基膦(2mmol)于室温下反应12h重结晶得到中间体4b;将中间体4b用甲苯(5ml)溶解之后与1-萘甲醛(1.861mmol)混合,加热到110℃的环境下反应24h后,减压除去溶剂得粗中间体5b,然后加入乙醇(5ml)溶液,同时加入硼氢化钠(1.508mmol),于室温下反应6h,然后将混合物用柱层析纯化,得到中间体6b;将三氯化磷(0.618mmol)滴加至二氯甲烷(3ml)中,降温至-78℃,然后将中间体6b溶解在二氯甲烷(2ml)中,用注射器缓慢加至反应液中,再加入吡啶(1.854mmol)在此温度下搅拌12h,移至室温滤出白色沉淀,减压除去溶剂后加入少量乙醚溶液,在低温下重结晶析出白色晶体7b(57%)。
实施例6
一种合成3,9-dichloro-5,7-bis(naphthalen-1-ylmethyl)-5,7-dihydrobenzo[d]benzo[4,5][1,3,2]diazaphospholo[1,2-a][1,3,2]diazaphosphole的方法,包括以下实验步骤:
在反应器中依次加入原料4-氯-2-叠氮溴苯2b(11mmol),溶剂DMSO(50ml),K2CO3(12mmol)和4-氯-2-叠氮苯胺1b(10mmol),将温度升至120℃反应36h。然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3b(9.4mmol,94%.);将3b(9.4mmol.)置于反应瓶内用甲苯(50ml)溶解,加入三苯基膦(20mmol)于室温下反应12h重结晶得到中间体4b(8.46mmol,90%);将中间体4b(8.46mmol)用甲苯(50ml)溶解之后与1-萘甲醛(18.61mmol)混合,加热到110℃的环境下反应24h后,减压除去溶剂得粗中间体5b(6.85mmol,81%),然后加入乙醇(50ml)溶液,同时加入硼氢化钠(15.08mmol),于室温下反应6h,然后将混合物用柱层析纯化,得到中间体6b(5.62mmol,82%);将三氯化磷(6.18mmol)滴加至二氯甲烷(30ml)中,降温至-78℃,然后将中间体6b(5.62mmol)溶解在二氯甲烷(20ml)中,用注射器缓慢加至反应液中,搅拌12h,移至室温滤出白色沉淀,减压除去溶剂后加入少量乙醚溶液,在低温下重结晶得到产品,经检测,未检测到所合成的产品,即目标收率为0%。
实施例7
一种合成3,9-dichloro-5,7-bis(naphthalen-1-ylmethyl)-5,7-dihydrobenzo[d]benzo[4,5][1,3,2]diazaphospholo[1,2-a][1,3,2]diazaphosphole的方法,包括以下实验步骤:
在反应器中依次加入原料4-氯-2-叠氮溴苯2b(11mmol),溶剂DMSO(50ml),K2CO3(12mmol)和4-氯-2-叠氮苯胺1b(10mmol),将温度升至120℃反应36h。然后缓慢加水淬灭反应,并用二氯甲烷萃取出有机相并用无水硫酸钠干燥,减压除去溶剂后得到橙色固体3b(9.4mmol,94%.);将3b(9.4mmol.)置于反应瓶内用甲苯(50ml)溶解,加入三苯基膦(20mmol)于室温下反应12h重结晶得到中间体4b(8.46mmol,90%);将中间体4b(8.46mmol)用甲苯(50ml)溶解之后与1-萘甲醛(18.61mmol)混合,加热到110℃的环境下反应24h后,减压除去溶剂得粗中间体5b(6.85mmol,81%),然后加入乙醇(50ml)溶液,同时加入氢气/钯碳,于室温下反应6h,然后将混合物用柱层析纯化,得到中间体6b(5.62mmol,82%);将三氯化磷(6.18mmol)滴加至二氯甲烷(30ml)中,降温至-78℃,然后将中间体6b(5.62mmol)溶解在二氯甲烷(20ml)中,用注射器缓慢加至反应液中,再加入吡啶(18.54mmol)在此温度下搅拌12h,移至室温滤出白色沉淀,减压除去溶剂后加入少量乙醚溶液,在低温下重结晶析出白色晶体7b(31%)。
实施例8
催化活性实验(催化Staudinger还原反应)
我们对所合成的新型四环稠合磷化物的催化活性进行了测试,主要测试了我们制备的新型四环稠合磷化物催化Staudinger还原反应的活性。测试结果如下:
反应条件:原料对硝基叠氮苯1mmol,溶剂5ml,磷化合物用量1.1mmol,反应时间为12小时。
此实验很好的说明了我们所制备的新型四环稠合磷化物对Staudinger还原反应是有效的。
上述的实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本申请中的实施例及实施例中的特征在不冲突的情况下,可以相互任意组合。本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围。即在此范围内的等同替换改进,也在本发明的保护范围之内。
一类四环稠合磷化物、合成方法及其催化应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。









动态评分
0.0