








专利摘要
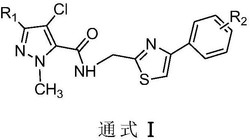
本发明涉及式Ⅰ所示N‑噻唑基吡啶甲酰胺衍生物及其药学上可接受的盐,药物组合物以及其在制备流感病毒神经氨酸酶抑制剂中的应用。R选自:甲基、乙基;R1选自:氢、3‑羟基、4‑羟基、3,4‑二羟基、3,5‑二羟基、2‑羟基‑3‑甲氧基、3‑羟基‑4‑甲氧基、4‑羟基‑3‑甲氧基;R2选自:H、3‑甲基、4‑甲基、5‑甲基或6‑甲基;或R2选自:H、2‑甲基、4‑甲基、5‑甲基或6‑甲基;或R2选自:H、2‑甲基、3‑甲基。
权利要求
1.一类化学结构式Ⅰ所示的N-噻唑基吡啶甲酰胺衍生物及其药学上可接受的盐:
式Ⅰ所示N-噻唑基吡啶甲酰胺衍生物选自式Ⅱ、Ⅲ或Ⅳ所示N-噻唑基吡啶甲酰胺衍生物:
其中,式Ⅱ、Ⅲ和Ⅳ中,R选自:甲基、乙基;R
Ⅱ式中,R
Ⅲ式中,R
Ⅳ式中,R
2.一类N-噻唑基吡啶甲酰胺衍生物及其药学上可接受的盐,选自下列化合物:
3.权利要求1所述的N-噻唑基吡啶甲酰胺衍生物的制备方法,其特征在于,它的制备反应如下:
式Ⅰ中,R、R
4.权利要求1或2所述的N-噻唑基吡啶甲酰胺衍生物及其在药学上可接受的盐在制备流感病毒神经氨酸酶抑制剂中的应用。
5.一种药物组合物,包括权利要求1或2中至少一种化合物和制药学上可用的载体。
说明书
技术领域
本发明涉及一类新化合物、其制备方法与应用,具体是N-噻唑基吡啶甲酰胺衍生物、其制备方法及其在制备流感病毒神经氨酸酶抑制剂的应用。
背景技术
Mills等[Bioorg Med Chem Lett,2010,20(1):87-91]描述了4-甲基-5-乙酰基-2-苯甲酰氨基噻唑1的制备方法,收率97%。经4-甲基-5-乙酰基-2-苯甲酰氨基噻唑制备的化合物2具有良好的抗囊性纤维化活性。
Koch课题组[Bioorg Med Chem Lett,2006,16(3):663-667]描述了新型噻唑类化合物3的合成,该类化合物对HIV病毒有很好的抑制活性。
Ian A.Yule等[Eur J Med Chem,2014,86:31-38]基于结构设计、合成了一系列抗菌剂,其中含有N-噻唑基吡啶甲酰胺结构的化合物4对大肠杆菌腺苷三磷酸酶具有抑制活性(IC500.42μM);在化合物4的基础上优化得到具有更高活性化合物5(IC50 0.098μM)。
Qun-Li Luo等[Bioorg Med Chem Lett,2005,15(3):635–638]描述了一系列含有吡啶-2-羧基噻唑-2-胺结构的甲硫氨酰氨基肽酶I类抑制剂,其中化合物6对于大肠杆菌甲硫氨酰氨基肽酶和鼠伤寒沙门氏菌甲硫氨酰氨基肽酶均具有抑制活性,其IC50分别为0.26μM和0.35μM。
世界专利[WO2013/082324A1,2012-11-29]描述了一系列噻唑酰胺类化合物的抗癌细胞增殖活性,发现具有N-噻唑基吡啶甲酰胺结构的化合物具有明显优越的抗癌细胞增殖活性。其中,化合物7对Hela细胞、K562细胞、MDA-MB-468细胞以及MDA-MB-231细胞均具有突出的抗增殖活性;化合物8对Hela细胞的抗增殖活性最优,其IC50值为8.3nM。
发明内容
本发明解决的技术问题是提供一类N-噻唑基吡啶甲酰胺衍生物、其制备方法、药物组合物和用途。
为解决本发明的技术问题,本发明提供如下技术方案:
本发明技术方案的第一方面是提供了一类如结构式Ⅰ所示N-噻唑基吡啶甲酰胺衍生物及其药学上可接受的盐:
式Ⅰ所示N-噻唑基吡啶甲酰胺衍生物选自式Ⅱ、Ⅲ或Ⅳ所示N-噻唑基吡啶甲酰胺衍生物:
其中,式Ⅱ、Ⅲ和Ⅳ中,R选自:甲基、乙基;R
Ⅱ式中,R
Ⅲ式中,R
Ⅳ式中,R
本发明技术方案的第一方面还提供的一类N-噻唑基吡啶甲酰胺衍生物选自下列化合物:
本发明技术方案的第二方面是提供了N-噻唑基吡啶甲酰胺衍生物的制备方法,其特征在于它的制备反应如下:
式Ⅰ所示N-噻唑基吡啶甲酰胺衍生物选自式Ⅱ、Ⅲ或Ⅳ所示N-噻唑基吡啶甲酰胺衍生物:
其中,式Ⅱ、Ⅲ和Ⅳ中,R选自:甲基、乙基;R
Ⅱ式中,R
Ⅲ式中,R
Ⅳ式中,R
本发明技术方案的第三方面是提供含有第一方面所述化合物及其药学上可接受的盐的药物组合物,该药物组合物含有治疗有效量的本发明的N-噻唑基吡啶甲酰胺衍生物及其药学上可接受的盐,以及任选的含有药用载体。其中所述的药用载体指药学领域常用的药用载体;该药物组合物可根据本领域公知的方法制备。可通过将本发明化合物及其药学上可接受的盐与一种或多种药学上可接受的固体或液体赋形剂和/或辅剂组合,制成适于人或动物使用的任何剂型。本发明化合物及其药学上可接受的盐在其药物组合物中的含量通常为0.1%~95%重量百分比。
本发明化合物及其药学上可接受的盐或含有它的药物组合物可以单位剂量形式给药,给药途径可为肠道或非肠道,如口服、静脉注射、肌肉注射、皮下注射、鼻腔、口腔粘膜、眼、肺和呼吸道、皮肤、阴道、直肠等。
给药剂型可以是液体剂型、固体剂型或半固体剂型。液体剂型可以是溶液剂(包括真溶液和胶体溶液)、乳剂(包括o/w型、w/o型和复乳)、混悬剂、注射剂(包括水针剂、粉针剂和输液)、滴眼剂、滴鼻剂、洗剂和搽剂等;固体剂型可以是片剂(包括普通片、肠溶片、含片、分散片、咀嚼片、泡腾片、口腔崩解片)、胶囊剂(包括硬胶囊、软胶囊、肠溶胶囊)、颗粒剂、散剂、微丸、滴丸、栓剂、膜剂、贴片、气(粉)雾剂、喷雾剂等;半固体剂型可以是软膏剂、凝胶剂、糊剂等。
本发明化合物及其药学上可接受的盐可以制成普通制剂、也制成是缓释制剂、控释制剂、靶向制剂及各种微粒给药系统。
为了将本发明化合物及其药学上可接受的盐制成片剂,可以广泛使用本领域公知的各种赋形剂,包括稀释剂、黏合剂、润湿剂、崩解剂、润滑剂、助流剂。稀释剂可以是淀粉、糊精、蔗糖、葡萄糖、乳糖、甘露醇、山梨醇、木糖醇、微晶纤维素、硫酸钙、磷酸氢钙、碳酸钙等;湿润剂可以是水、乙醇、异丙醇等;粘合剂可以是淀粉浆、糊精、糖浆、蜂蜜、葡萄糖溶液、微晶纤维素、阿拉伯胶浆、明胶浆、羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、乙基纤维素、丙烯酸树脂、卡波姆、聚乙烯吡咯烷酮、聚乙二醇等;崩解剂可以是干淀粉、微晶纤维素、低取代羟丙基纤维素、交联聚乙烯吡咯烷酮、交联羧甲基纤维素钠、羧甲基淀粉钠、碳酸氢钠与枸橼酸、聚氧乙烯山梨糖醇脂肪酸酯、十二烷基磺酸钠等;润滑剂和助流剂可以是滑石粉、二氧化硅、硬脂酸盐、酒石酸、液体石蜡、聚乙二醇等。
还可以将片剂进一步制成包衣片,例如糖包衣片、薄膜包衣片、肠溶包衣片,或双层片和多层片。
为了将给药单元制成胶囊剂,可以将有效成分本发明化合物及其药学上可接受的盐与稀释剂、助流剂混合,将混合物直接置于硬胶囊或软胶囊中。也可将有效成分本发明化合物及其药学上可接受的盐先与稀释剂、黏合剂、崩解剂制成颗粒或微丸,再置于硬胶囊或软胶囊中。用于制备本发明化合物及其药学上可接受的盐片剂的各稀释剂、黏合剂、润湿剂、崩解剂、助流剂品种也可用于制备本发明化合物及其药学上可接受的盐的胶囊剂。
为将本发明化合物及其药学上可接受的盐制成注射剂,可以用水、乙醇、异丙醇、丙二醇或它们的混合物作溶剂并加入适量本领域常用的增溶剂、助溶剂、pH调剂剂、渗透压调节剂。增溶剂或助溶剂可以是泊洛沙姆、卵磷脂、羟丙基-β-环糊精等;pH调剂剂可以是磷酸盐、醋酸盐、盐酸、氢氧化钠等;渗透压调节剂可以是氯化钠、甘露醇、葡萄糖、磷酸盐、醋酸盐等。如制备冻干粉针剂,还可加入甘露醇、葡萄糖等作为支撑剂。
此外,如需要,也可以向药物制剂中添加着色剂、防腐剂、香料、矫味剂或其它添加剂。
为达到用药目的,增强治疗效果,本发明的药物或药物组合物可用任何公知的给药方法给药。
本发明技术方案的第四方面是提供本发明所述N-噻唑基吡啶甲酰胺衍生物及其药学上可接受的盐以及第三方面所述药物组合物在制备流感病毒神经氨酸酶抑制剂方面的应用。
有益技术效果:
本发明的N-噻唑基吡啶甲酰胺衍生物是一类对流感病毒神经氨酸酶具有抑制活性的化合物。
具体实施方式
以下实施例旨在说明本发明而不是对本发明的进一步限定。
实施例1
4-甲基-5-[3-(4-羟基苯基)丙烯酰基]-2-苯甲酰氨基噻唑(A)的制备
(1)4-甲基-5-乙酰基-2-苯甲酰氨基噻唑的制备
6.0mmol苯甲酸溶于10mL DMF溶液中,分批加入0.972g(6.0mmol)N,N'-羰基二咪唑(CDI),室温搅拌30min,加入4.0mmol 4-甲基-5-乙酰基-2-氨基噻唑,85℃反应8h。冷至室温,反应液倒入30mL冰水,析出大量固体,粗品经乙酸乙酯/四氢呋喃重结晶得白色固体4-甲基-5-乙酰基-2-苯甲酰氨基噻唑,收率61.0%,m.p.228~230℃。
或按文献[Bioorg Med Chem Lett,2010,20(1):87-91]方法制备:
(2)4-甲基-5-[3-(4-羟基苯基)丙烯酰基]-2-苯甲酰氨基噻唑(A)的制备
1.0mmol 4-甲基-5-乙酰基-2-苯甲酰氨基噻唑与1.0mmol对羟基苯甲醛溶液于6.0mL乙醇溶液中,通入干燥HCl气体至饱和,室温反应6h,粗品经乙醇重结晶得黄色固体4-甲基-5-[3-(4-羟基苯基)丙烯酰基]-2-苯甲酰氨基噻唑A,收率67.7%,m.p.262~264℃。
实施例2
4-甲基-5-[3-(4-羟基苯基)丙烯酰基]-2-(3-吡啶甲酰氨基)噻唑(C)的合成
(1)N-[4-甲基-5-乙酰基噻唑-2-基]-3-吡啶甲酰胺的合成
按实施例1的方法,6.0mmol烟酸与4.0mmol 4-甲基-5-乙酰基-2-氨基噻唑反应8h,粗品经乙酸乙酯/四氢呋喃重结晶得黄色固体N-[4-甲基-5-乙酰基噻唑-2-基]-3-吡啶甲酰胺,收率55.7%,m.p.226~228℃。
(2)N-[4-甲基-5-[3-(4-羟基苯基)丙烯酰基]噻唑-2-基]-3-吡啶甲酰胺(C)的合成
按实施例1的方法,1.0mmol N-[4-甲基-5-乙酰基噻唑-2-基]-3-吡啶甲酰胺与1.0mmol对羟基苯甲醛反应6h,粗品经乙醇重结晶得黄色固体N-[4-甲基-5-[3-(4-羟基苯基)丙烯酰基]噻唑-2-基]-3-吡啶甲酰胺C,收率67.7%,m.p.243~245℃。
实施例3
N-噻唑基吡啶甲酰胺衍生物的抗流感病毒神经氨酸酶活性
1.实验原理
化合物MUNANA是神经氨酸酶的特异性底物,在神经氨酸酶作用下产生的代谢产物在360nm照射激发下,可以产生450nm荧光,荧光强度的变化可以灵敏地反应神经氨酸酶活性。酶都来自A/PR/8/34(H1N1)病毒毒株。
2.实验方法
在酶反应体系中,一定浓度样品与流感病毒神NA悬浮于反应缓冲液中(pH 6.5),加入荧光底物MUNANA启动反应体系,37℃孵育40分钟后,加反应终止液终止反应。在激发波长360nm和发射波长为450nm的参数条件下,测定荧光强度值。反应体系的荧光强度可以反映酶的活性。根据荧光强度的减少量可以计算化合物对NA活性的抑制率。
3.检测样品:实施例化合物
4.活性结果
化合物A和C在反应系统中检测浓度40.0μg/mL时对神经氨酸酶的抑制率和IC50值列入表1。
表1化合物A和C对神经氨酸酶H1N1的抑制活性和IC50
N-噻唑基吡啶甲酰胺衍生物具有抗流感病毒神经氨酸酶活性,可用于制备流感病毒神经氨酸酶抑制剂。
化合物C对神经氨酸酶H1N1的抑制活性高于化合物A的活性。
N-噻唑基吡啶甲酰胺衍生物及其制备方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


![N-[5-(1,2,4-三唑-1-基)噻唑-2-基]吗啉基酰胺及其医药用途](https://www.zhichawang.com/images/CN107286147A/CN107286147A.jpg)






动态评分
0.0