专利摘要
本发明公开了一种去氢枞酸噁唑烷酮衍生物及其制备方法和应用,制备时先将去氢枞酸与卤代环氧丙烷反应制备去氢枞酸缩水甘油酯;将上步将制备好的去氢枞酸缩水甘油酯与芳香伯胺反应,制备3'‑芳香胺基‑2'‑羟基去氢枞酸丙酯;然后将3'‑芳香胺基‑2'‑羟基去氢枞酸丙酯与环合试剂反应制备去氢枞酸噁唑烷酮衍生物。制备的去氢枞酸噁唑烷酮衍生物作为抗肿瘤化合物的应用。与现有技术相比,本发明提供了一类新的去氢枞酸噁唑烷酮衍生物的制备方法,其制备周期短,操作简单,成本低,且得到的衍生物纯度高,质量稳定;申请人还发现,通过在去氢枞酸骨架上引入功能性基团噁唑烷酮可以改善化合物的抗肿瘤活性,可作为抗肿瘤化合物。
权利要求
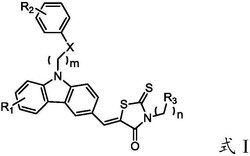
1.去氢枞酸噁唑烷酮衍生物,其特征是,具有下述式(I)所示结构:
其中,
R为芳香基,具体为苯基、邻甲苯基、间甲苯基、对甲苯基、邻甲氧基苯基、对甲氧基苯基、邻氟苯基、间氟苯基、对氟苯基、间氯苯基、对氯苯基、间溴苯基、对溴苯基、间乙炔基苯基。
2.根据权利要求1所述的去氢枞酸噁唑烷酮衍生物的制备方法,其特征是,通式(I)所示结构的去氢枞酸噁唑烷酮衍生物的合成反应式如下:
制备具体步骤如下:
(A)先将去氢枞酸与卤代环氧丙烷反应制备去氢枞酸缩水甘油酯;
(B)将上步制备好的去氢枞酸缩水甘油酯与芳香伯胺反应,制备3'-芳香胺基-2'-羟基去氢枞酸丙酯;
(C)然后将3'-芳香胺基-2'-羟基去氢枞酸丙酯与环合试剂反应制备去氢枞酸噁唑烷酮衍生物;
其中,
R为芳香基,具体为苯基、邻甲苯基、间甲苯基、对甲苯基、邻甲氧基苯基、对甲氧基苯基、邻氟苯基、间氟苯基、对氟苯基、间氯苯基、对氯苯基、间溴苯基、对溴苯基、间乙炔基苯基。
3.根据权利要求2所述的制备方法,其特征是:步骤(A)所述制备去氢枞酸缩水甘油酯,还可将去氢枞酸与环氧氯丙烷反应,或者去氢枞酸与环氧溴丙烷反应;
所述制备去氢枞酸缩水甘油酯反应在缚酸剂条件下进行,缚酸剂为无水碳酸钠,无水碳酸钾,碳酸铯,三乙胺,吡啶中的一种;
所述制备去氢枞酸缩水甘油酯反应在惰性溶剂中进行,惰性有机溶剂选自苯,四氢呋喃,二氧六环,乙腈,丙酮,乙酸乙酯,二氯甲烷,三氯甲烷;
所述制备去氢枞酸缩水甘油酯反应温度:选择室温至溶剂回流温度。
4.根据权利要求3所述的制备方法,其特征是:步骤(A)所述制备去氢枞酸缩水甘油酯,是将去氢枞酸与环氧溴丙烷反应,去氢枞酸与环氧溴丙烷的摩尔比是1:1.1;
所述缚酸剂为无水碳酸钾;
所述惰性有机为乙腈或丙酮;
所述反应温度为40-80℃。
5.根据权利要求2所述的制备方法,其特征是:步骤(B)所述去氢枞酸缩水甘油酯与芳香伯胺类化合物的摩尔比1:1.2;
所述芳香伯胺是苯胺、邻甲苯胺、间甲苯胺、对甲苯胺、邻甲氧基苯胺、对甲氧基苯胺、邻氟苯胺、间氟苯胺、对氟苯胺、间氯苯胺、对氯苯胺、间溴苯胺、对溴苯胺、间氨基苯乙炔;
所述去氢枞酸缩水甘油酯与芳香伯胺反应在催化剂条件下进行,催化剂为高氯酸镁或高氯酸锌;
所述去氢枞酸缩水甘油酯与芳香伯胺反应在有机溶剂中进行,有机溶剂选自甲醇,乙醇,异丙醇,四氢呋喃,二氧六环,乙腈,丙酮,乙酸乙酯,二氯甲烷,三氯甲烷;
所述去氢枞酸缩水甘油酯与芳香伯胺反应温度:选择室温至溶剂回流温度。
6.根据权利要求5所述的制备方法,其特征是:所述去氢枞酸缩水甘油酯与芳香伯胺反应温度为60-80℃。
7.根据权利要求2所述的制备方法,其特征是:步骤(C)所述环合反应通常在常规反应器中进行,以薄层层析跟踪检测环合反应是否完全,反应至完全需要1~2h的时间。
8.根据权利要求2所述的制备方法,其特征是:步骤(C)所述环合试剂为光气,三光气,N,N′-羰基二咪唑,碳酸二甲酯,碳酸二乙酯中的一种;
制备去氢枞酸噁唑烷酮衍生物的反应在催化剂条件下进行,催化剂为无水碳酸钾,无水碳酸钠,氢氧化钾,氢氧化钠中的一种;
制备去氢枞酸噁唑烷酮衍生物的反应在惰性溶剂中进行,惰性溶剂选自苯,四氢呋喃,二氧六环,乙腈,丙酮,乙酸乙酯,二氯甲烷,三氯甲烷中的一种或多种;
制备去氢枞酸噁唑烷酮衍生物的反应温度:选择0-30℃至溶剂回流温度。
9.根据权利要求8所述的制备方法,其特征是:所述环合试剂为三光气,去氢枞酸丙酯衍生物与三光气的摩尔比为3:1。
说明书
技术领域
本发明涉及医药技术领域,具体涉及去氢枞酸噁唑烷酮衍生物及其制备方法和应用。
背景技术
癌症是当今危害人类健康最为严重的疾病之一,而且发病率呈上升趋势,然而目前临床上抗肿瘤药物,大多数存在毒性大、效果不佳、易产生多药耐药等缺点,因此研发高效低毒的新型抗肿瘤药物是亟待解决的重大问题。
去氢枞酸又名脱氢松香酸或脱氢枞酸,是从歧化松香中分离出的一种天然的三环二萜类树脂酸,具有与许多活性天然化合物相似或相近的结构骨架,也有良好的生物相容性和生物可降解性,其本身具有抗炎、抗菌、抗肿瘤等多种生物活性,是一个极具潜力的先导化合物。同时,大部分去氢枞酸衍生物具有抗菌、抗肿瘤、抗病毒、除草、杀虫等生物活性(王秀,庞富华,李芳耀.天然产物研究与开发,2017,29(9):1621-1625.),因此对去氢枞酸的结构修饰成为药物研究的热点领域之一。尤其是,研究发现去氢枞酸衍生物对宫颈癌、肝癌、肺癌、前列腺癌、卵巢癌和乳腺癌细胞株有较强抑制增殖活性。而噁唑烷酮类是抗菌、抗病毒作用的重要潜在药效团。例如:2000年上市的第一个噁唑烷酮类抗菌药利奈唑胺对甲氧西林敏感或耐药葡萄球菌、万古霉素敏感或耐药肠球菌、青霉素敏感或耐药肺炎链球菌均显示了良好的抗菌作用。(Moellering,R.C.Ann.Intern.Med,2003,138(2):135-142)。故我们以去氢枞酸为活性母核,拟引进噁唑烷酮药效团,以拼合二者的活性,研发出新型低毒高效抗癌化合物。
虽然现有文献中报道了许多在去氢枞酸母核上进行结构改造,得到一系列去氢枞酸衍生物,但并未见有去氢枞酸与噁唑烷酮拼合得到去氢枞酸噁唑烷酮类衍生物的公开报道。
发明内容
本发明要解决的技术问题是提供一类通过在去氢枞酸羧基结构上引入药效基团噁唑烷酮得到的去氢枞酸噁唑烷酮衍生物及其制备方法和应用。
本发明所述的去氢枞酸噁唑烷酮衍生物,具有下述式(I)所示结构:
其中,R为芳香基,具体为苯基、邻甲苯基、间甲苯基、对甲苯基、邻甲氧基苯基、对甲氧基苯基、邻氟苯基、间氟苯基、对氟苯基、间氯苯基、对氯苯基、间溴苯基、对溴苯基、间乙炔基苯基等。
上述通式(I)所示结构的去氢枞酸噁唑烷酮衍生物的合成反应式如下:
制备具体步骤如下:
(A)先将去氢枞酸与卤代环氧丙烷反应制备去氢枞酸缩水甘油酯;
(B)将上步将制备好的去氢枞酸缩水甘油酯与芳香伯胺反应,制备3'-芳香胺基-2'-羟基去氢枞酸丙酯;
(C)然后将3'-芳香胺基-2'-羟基去氢枞酸丙酯与环合试剂反应制备去氢枞酸噁唑烷酮衍生物。
上述步骤(A)所述制备去氢枞酸缩水甘油酯,还可将去氢枞酸与环氧氯丙烷反应,或者去氢枞酸与环氧溴丙烷反应,优选去氢枞酸与环氧溴丙烷反应;
反应优选在缚酸剂条件下进行,优选的缚酸剂有无水碳酸钠,无水碳酸钾、碳酸铯,三乙胺,吡啶,更优选无水碳酸钾;
反应优选在惰性溶剂中进行,惰性有机溶剂选自苯,四氢呋喃,二氧六环,乙腈,丙酮,乙酸乙酯,二氯甲烷,三氯甲烷,更优选丙酮或乙腈。
反应温度:选择室温至溶剂回流温度,优选40-80℃。
上述步骤(B)所述将制备好的去氢枞酸缩水甘油酯与芳香伯胺反应,制备3'-芳香胺基-2'-羟基去氢枞酸丙酯;
所述芳香伯胺具体可以是苯胺、邻甲苯胺、间甲苯胺、对甲苯胺、邻甲氧基苯胺、对甲氧基苯胺、邻氟苯胺、间氟苯胺、对氟苯胺、间氯苯胺、对氯苯胺、间溴苯胺、对溴苯胺、间氨基苯乙炔等;
反应优选在催化剂条件下进行,优选的催化剂有高氯酸镁、高氯酸锌;
反应优选在有机溶剂中进行,有机溶剂选自甲醇,乙醇,异丙醇,四氢呋喃,二氧六环,乙腈,丙酮,乙酸乙酯,二氯甲烷,三氯甲烷,更优选甲醇或乙醇;
反应温度:选择室温至溶剂回流温度,优选60-80℃。
上述步骤(C)所述将3'-芳香胺基-2'-羟基去氢枞酸丙酯与环合试剂反应制备去氢枞酸噁唑烷酮衍生物;
所述环合试剂有光气,三光气,N,N′-羰基二咪唑,碳酸二甲酯,碳酸二乙酯,优选三光气;
制备去氢枞酸噁唑烷酮衍生物的反应优选在催化剂条件下进行,优选的催化剂有无水碳酸钾,无水碳酸钠,氢氧化钾,氢氧化钠,更优选氢氧化钠;
制备去氢枞酸噁唑烷酮衍生物的反应优选在惰性溶剂中进行,惰性溶剂选自苯,四氢呋喃,二氧六环,乙腈,丙酮,乙酸乙酯,二氯甲烷,三氯甲烷中的一种或多种,更优选二氯甲烷或四氢呋喃;
制备去氢枞酸噁唑烷酮衍生物的反应温度:选择0℃至溶剂回流温度,优选0-30℃。
所述的环合反应通常在常规反应器中进行,以薄层层析跟踪检测环合反应是否完全,反应至完全需要1~2h的时间。
所述去氢枞酸与环氧溴丙烷的摩尔比优选是1:1.1,去氢枞酸缩水甘油酯与芳香伯胺类化合物的摩尔优选比1:1.2,环合试剂优选三光气,去氢枞酸丙酯衍生物与三光气摩尔优选比3:1,所述芳香伯胺类化合物的用量超过上述范围反应也同样可以生成目标产物,后序的分离难度增加。
本发明还包括上述通式(I)所示结构的去氢枞酸噁唑烷酮衍生物作为抗肿瘤化合物的应用。
与现有技术相比,本发明提供了一类新的去氢枞酸噁唑烷酮衍生物的制备方法,其制备周期短,操作简单,成本低,且得到的衍生物纯度高,质量稳定;申请人还发现,通过在去氢枞酸骨架上引入功能性基团噁唑烷酮可以改善化合物的抗肿瘤活性,可作为抗肿瘤化合物。
具体实施方式
下面以具体实施例对本发明内容作进一步的说明,但本发明并不局限于这些实施例范围。
在以下各实施例中,去氢枞酸用化合物1表示,去氢枞酸缩水甘油酯用化合物2表示,3'-芳香胺基-2'-羟基去氢枞酸丙酯用化合物3表示。去氢枞酸噁唑烷酮衍生物用4表示
实施例1:去氢枞酸缩水甘油酯(化合物2)的制备
称量化合物1(9g,29.95mmol)、环氧溴丙烷(4.10g,32.5mmol)和无水碳酸钾(9.4g,1.39mmol)溶于40ml丙酮置于圆底烧瓶中,在温度为60℃下、回流反应至完全(TLC跟踪检测,约4h),自然冷却至室温后,抽滤,用乙酸乙酯多次洗涤,合并滤液,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到淡黄透明油状液体8.85g,产率82.94%。
因此,上述化合物2为去氢枞酸缩水甘油酯,其结构式如下式所示:
实施例2:3'-苯胺基-2'-羟基去氢枞酸丙酯(化合物3a)的制备的制备
称量化合物2(120mg,0.34mmol)、苯胺(37.99mg,0.41mmol)和六合水高氯酸锌(5mg,)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层用无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到白色固体3a 112.53mg,产率74.35%。1H NMR(600MHz,DMSO)δ7.16(d,J=8.2Hz,1H),7.01(s,1H),6.97(d,J=7.4Hz,2H),6.81(s,1H),6.56(d,J=3.9Hz,2H),6.50(s,1H),5.13(s,1H),4.07(s,1H),3.97(s,1H),3.84(s,1H),3.11(s,1H),3.00(s,1H),2.81–2.72(m,3H),2.50(s,1H),2.28(s,1H),2.12(s,1H),1.73(t,J=8.8Hz,3H),1.64(s,1H),1.58(s,1H),1.33(d,J=11.8Hz,2H),1.21(s,3H),1.15(d,J=3.7Hz,3H),1.14(d,J=3.7Hz,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ177.58,148.70,146.66,145.15,134.21,128.91,126.54,124.18,123.82,115.79,112.11,67.02,66.92,66.64,66.58,47.17,46.36,44.77,37.73,36.57,36.19,32.96,29.63,24.99,23.98,21.28,21.25,18.18,16.39.
因此,可确定上述化合物为3a 3'-苯胺基-2'-羟基去氢枞酸丙酯产物,其结构式如下式所示:
实施例3:3'-邻甲苯胺基-2'-羟基去氢枞酸丙酯(化合物3b)的制备
称量化合物2(120mg,0.34mmol)、邻甲苯胺(43.71mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到白色固体3b 125.63mg,产率80.49%。1H NMR(600MHz,DMSO)δ7.14(s,1H),7.00–6.89(m,3H),6.80(s,1H),6.48(d,J=7.3Hz,2H),5.16(s,1H),4.68(s,1H),4.07(s,1H),4.00(s,1H),3.88(s,1H),3.16(s,1H),3.03(s,1H),2.80–2.72(m,3H),2.49(s,1H),2.27(s,1H),2.11(s,1H),2.04(s,2H),1.75–1.70(m,3H),1.62(s,1H),1.57(s,1H),1.32(d,J=12.2Hz,2H),1.20(s,3H),1.13(dd,J=6.9,3.6Hz,6H),1.11(s,3H).13C NMR((151MHz,DMSO)δ178.07,147.17,146.78,145.63,134.70,130.34,127.25,127.04,124.66,124.31,122.29,116.41,109.64,67.32,67.24,47.66,47.06,46.97,45.28,38.22,37.07,36.69,33.45,30.13,25.48,24.48,21.74,18.68,18.05,16.89.
因此,可确定上述化合物为3b 3'-邻甲苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例4:3'-间甲苯胺基-2'-羟基去氢枞酸丙酯(化合物3c)的制备
称量化合物2(120mg,0.34mmol)、间甲苯胺(43.71mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到淡黄色油状液体3c108.58mg,产率69.29%。1H NMR(600MHz,DMSO)δ7.15(s,1H),6.97(s,1H),6.89(s,1H),6.80(s,1H),6.41–6.30(m,3H),5.39(s,1H),5.11(s,1H),4.06(s,1H),3.98(s,1H),3.82(s,1H),3.09(s,1H),2.98(s,1H),2.80–2.71(m,3H),2.50(s,1H),2.28(s,1H),2.13(s,2H),1.98(s,1H),1.73(t,J=10.8Hz,3H),1.63(s,1H),1.58(s,1H),1.33(d,J=12.5Hz,2H),1.21(s,3H),1.15(d,J=2.8Hz,3H),1.14(d,J=2.8Hz,3H),1.12(s,3H).13C NMR(151MHz,DMSO)δ177.58,148.72,146.66,145.15,137.90,134.20,128.78,126.52,124.20,123.83,116.74,114.22,112.72,109.52,66.97,66.59,47.17,46.31,44.81,37.71,36.58,36.18,32.95,29.65,25.02,23.97,21.39,18.18,16.40,14.14.
因此,可确定上述化合物3c为3'-间甲苯胺基-2'-羟基去氢枞酸丙酯(化合物3c),其结构式如下式所示:
实施例5:3'-对甲苯胺基-2'-羟基去氢枞酸丙酯(化合物3d)的制备
称量化合物2(120mg,0.34mmol)、对甲苯胺(43.71mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到白色固体3d 114.65mg,产率73.46%。1H NMR(600MHz,DMSO)δ7.15(s,1H),6.97(s,1H),6.85–6.79(m,3H),6.48(d,J=6.7Hz,2H),5.29(s,1H),5.11(s,1H),4.06(s,1H),3.96(s,1H),3.82(s,1H),3.08(s,1H),2.97(s,1H),2.81–2.73(m,3H),2.50(s,1H),2.28(s,1H),2.12(s,2H),2.11(s,1H),1.81–1.68(m,3H),1.63(s,1H),1.57(s,1H),1.33(d,J=11.4Hz,2H),1.21(s,3H),1.15(s,3H),1.14(d,J=1.5Hz,3H),1.12(s,3H).13C NMR(151MHz,DMSO)δ177.58,148.72,146.66,145.15,137.90,134.20,128.78,126.52,124.20,123.83,116.74,114.22,112.72,109.52,66.97,66.59,47.17,46.31,44.81,37.71,36.58,36.18,32.95,29.65,25.02,23.97,21.39,18.18,16.40,14.14.
因此,可确定上述化合物3d为3'-对甲苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例6:3'-邻甲氧基苯胺基-2'-羟基去氢枞酸丙酯3e的制备
称量化合物2(120mg,0.34mmol)、邻甲氧基苯胺(50.49mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到淡黄色油状液体3e104.29mg,产率64.59%。1H NMR(600MHz,DMSO)δ7.15(s,1H),6.98(s,1H),6.80(d,J=13.7Hz,2H),6.71(s,1H),6.54(d,J=18.5Hz,2H),5.35–4.87(m,1H),4.06(dq,J=11.0,5.7Hz,1H),4.01–3.94(m,1H),3.92–3.83(m,1H),3.76(s,3H),3.16(s,1H),2.99(s,1H),2.81–2.73(m,3H),2.50(s,1H),2.31(s,1H),2.12(s,1H),1.73(t,J=10.6Hz,3H),1.63(s,1H),1.58(s,1H),1.33(s,2H),1.21(s,3H),1.16(s,3H),1.15(s,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ177.53,146.68,145.15,134.20,126.54,124.17,123.82,121.05,109.86,66.86,66.77,66.65,55.34,47.19,46.24,44.76,37.71,36.59,36.18,32.96,29.63,24.99,23.97,21.27,18.18,16.39.
因此,可确定上述化合物3e为3'-邻甲氧基苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例7:3'-对甲氧基苯胺基-2'-羟基去氢枞酸丙酯(化合物3f)的制备
称量化合物2(120mg,0.34mmol)、对甲氧基苯胺(50.49mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到无色油状液体3f114.33mg,产率70.81%。1H NMR(600MHz,DMSO)δ7.15(s,1H),6.98(s,1H),6.82(s,1H),6.67(d,J=4.1Hz,2H),6.55(d,J=8.7Hz,2H),5.11(s,1H),4.07(s,1H),3.96(s,1H),3.83(s,1H),3.60(t,J=17.6Hz,3H),3.06(s,1H),2.96(s,1H),2.83–2.69(m,3H),2.53–2.47(m,1H),2.29(s,1H),2.11(s,1H),1.73(t,J=9.8Hz,3H),1.63(s,1H),1.58(s,1H),1.33(d,J=12.3Hz,2H),1.21(s,3H),1.15(s,3H),1.14(s,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ177.54,151.12,146.68,145.15,134.21,126.53,124.18,123.82,114.64,66.98,66.70,55.35,47.44,47.16,44.78,37.71,36.58,36.18,32.95,29.63,24.98,23.97,21.26,18.18,16.40.
因此,可确定上述化合物3f为3'-对甲氧基苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例8:3'-邻氟苯胺基-2'-羟基去氢枞酸丙酯(化合物3g)的制备
称量化合物2(120mg,0.34mmol)、邻氟苯胺(45.56mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到无色油状液体3g124.53mg,产率78.32%。1H NMR(600MHz,DMSO)δ7.15(s,1H),6.97(d,J=6.2Hz,2H),6.83(d,J=15.7Hz,2H),6.70(s,1H),6.52(s,1H),5.21(s,1H),4.09(s,1H),3.99(s,1H),3.88(s,1H),3.59(s,1H),3.05(s,1H),2.82–2.72(m,3H),2.50(s,1H),2.29(s,1H),2.11(s,1H),1.80–1.68(m,3H),1.63(s,1H),1.58(s,1H),1.33(d,J=10.5Hz,2H),1.23–1.18(m,3H),1.15(dd,J=6.9,3.2Hz,6H),1.13(s,3H).13C NMR(151MHz,DMSO)δ178.05,152.24,150.67,147.17,145.64,137.19,134.70,127.03,125.24,124.31,116.12,114.92,112.59,68.95,67.33,66.44,47.68,46.58,45.27,38.21,37.07,33.44,30.11,25.48,24.48,21.72,18.67,16.89,15.59.
因此,可确定上述化合物3g为3'-邻氟苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例9:3'-间氟苯胺基-2'-羟基去氢枞酸丙酯(化合物3h)的制备
称量化合物2(120mg,0.34mmol)、间氟苯胺(45.56mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到淡黄色油状液体3h116.34mg,产率73.17%。1H NMR(600MHz,DMSO)δ7.15(s,1H),6.99(d,J=13.0Hz,2H),6.81(s,1H),6.30(d,J=59.6Hz,2H),5.94(s,1H),5.14(s,1H),4.02(d,J=60.3Hz,2H),3.82(s,1H),3.11(s,1H),3.01(s,1H),2.81–2.71(m,3H),2.50(s,1H),2.28(s,1H),1.98(s,1H),1.73(d,J=14.7Hz,3H),1.64(s,1H),1.58(s,1H),1.32(s,2H),1.21(s,3H),1.15(s,3H),1.14(s,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ177.57,164.38,162.79,150.97,146.66,145.15,134.20,130.16,126.52,124.18,123.82,108.36,101.68,98.08,66.84,66.49,65.92,47.18,46.17,44.80,37.70,36.58,32.95,29.64,23.97,21.26,18.18,16.39,15.10.
因此,可确定上述化合物3h为3'-间氟苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例10:3'-对氟苯胺基-2'-羟基去氢枞酸丙酯(化合物3i)的制备
称量化合物2(120mg,0.34mmol)、对氟苯胺(45.56mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到无色油状液体3i115.32mg,产率72.53%。1H NMR(600MHz,DMSO)δ7.17(s,1H),6.98(s,1H),6.86–6.78(m,3H),6.54(d,J=8.8Hz,2H),5.48(s,1H),4.07(s,1H),3.96(s,1H),3.82(s,1H),3.08(s,1H),2.96(s,1H),2.76(t,J=8.9Hz,3H),2.50(s,1H),2.29(s,1H),1.98(s,1H),1.73(t,J=8.7Hz,3H),1.64(s,1H),1.57(s,1H),1.35(d,J=42.3Hz,2H),1.20(s,3H),1.16(s,3H),1.14(d,J=2.3Hz,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ177.54,155.06,153.53,146.66,145.52,145.16,134.20,126.53,124.18,123.84,115.29,115.15,112.79,67.01,66.92,66.61,47.19,44.78,37.73,36.57,36.16,32.95,29.64,24.99,23.96,21.23,18.18,16.39,14.14.
因此,可确定上述化合物3i为3'-对氟苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例11:3'-间氯苯胺基-2'-羟基去氢枞酸丙酯(化合物3j)的制备
称量化合物2(120mg,0.34mmol)、间氯苯胺(52.30mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到无色油状液体3j123.45mg,产率75.01%。1H NMR(600MHz,DMSO)δ7.15(s,1H),6.99(d,J=17.2Hz,2H),6.81(s,1H),6.59(s,1H),6.51(d,J=8.1Hz,2H),5.94(s,1H),4.07(s,1H),3.96(s,1H),3.82(s,1H),3.10(s,1H),3.00(s,1H),2.83–2.72(m,3H),2.50(s,1H),2.28(s,1H),2.11(s,1H),1.74(t,J=10.2Hz,3H),1.63(s,1H),1.58(s,1H),1.33(s,2H),1.21(s,3H),1.15(s,3H),1.14(s,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ177.56,150.35,146.66,145.15,134.20,133.73,130.34,126.53,124.18,123.83,115.02,111.16,110.76,66.85,66.50,47.19,46.05,44.82,37.71,36.59,36.21,32.95,29.65,25.01,23.97,21.24,18.18,16.41.
因此,可确定上述化合物3j为3'-间氯苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例12:3'-对氯苯胺基-2'-羟基去氢枞酸丙酯(化合物3k)的制备
称量化合物2(120mg,0.34mmol)、对氯苯胺(52.30mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到无色油状液体3k115.52mg,产率70.19%。1H NMR(600MHz,DMSO)δ7.16(d,J=8.1Hz,1H),6.99(d,J=16.9Hz,3H),6.81(s,1H),6.56(d,J=8.7Hz,2H),5.75(s,1H),5.11(s,1H),4.09(s,1H),3.94(s,1H),3.83(s,1H),3.09(s,1H),2.83–2.67(m,3H),2.50(s,1H),2.29(s,1H),2.10(s,1H),1.74(t,J=12.1Hz,3H),1.63(s,1H),1.58(s,1H),1.32(s,2H),1.21(s,3H),1.16(s,3H),1.15(s,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ178.02,148.25,147.17,145.66,134.69,129.05,127.02,124.66,124.32,119.40,113.89,67.46,67.04,47.66,46.77,45.29,38.23,37.07,36.65,33.44,30.09,25.46,24.46,21.72,18.67,16.89.
因此,可确定上述化合物3k为3'-对氯苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例13:3'-间溴苯胺基-2'-羟基去氢枞酸丙酯(化合物3l)的制备
称量化合物2(120mg,0.34mmol)、间溴苯胺(70.52mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到无色油状液体3l124..23mg,产率69.13%。1H NMR(600MHz,DMSO)δ7.15(s,1H),6.96(d,J=19.4Hz,2H),6.82(s,1H),6.74(s,1H),6.62(s,1H),6.55(s,1H),5.95(s,1H),4.06(s,1H),3.96(s,1H),3.81(dd,J=10.3,5.1Hz,1H),3.10(s,1H),3.00(s,1H),2.83–2.73(m,3H),2.50(s,1H),2.31(s,1H),2.11(s,1H),1.74(t,J=10.6Hz,3H),1.63(s,1H),1.58(s,1H),1.33(d,J=4.6Hz,2H),1.21(s,3H),1.15(s,3H),1.14(s,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ177.56,150.51,146.67,145.15,134.21,130.66,126.53,124.19,123.83,122.45,117.91,114.07,111.13,66.89,66.49,47.20,46.02,44.82,37.71,36.59,36.18,32.95,29.66,25.01,23.98,21.28,18.19,16.42.
因此,可确定上述化合物3l为3'-间溴苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例14:3'-对溴苯胺基-2'-羟基去氢枞酸丙酯(化合物3m)的制备
称量化合物2(120mg,0.34mmol)、对溴苯胺(70.52mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到无色油状液体3m121.42mg,产率67.56%。1H NMR(600MHz,DMSO)δ7.15(s,1H),7.11(d,J=8.5Hz,2H),6.97(s,1H),6.80(s,1H),6.51(d,J=8.5Hz,2H),5.80(s,1H),4.06(s,1H),3.95(s,1H),3.80(s,1H),3.08(s,1H),2.98(s,1H),2.80–2.72(m,3H),2.50(s,1H),2.28(s,1H),2.09(s,1H),1.71(d,J=10.8Hz,3H),1.63(s,1H),1.57(s,1H),1.32(d,J=11.8Hz,2H),1.20(s,3H),1.15(s,3H),1.14(s,3H),1.12(s,3H).13C NMR(151MHz,DMSO)δ177.56,148.07,146.66,145.16,134.18,131.36,126.52,124.17,123.83,113.97,106.24,66.83,66.52,47.18,46.24,46.15,44.77,37.71,36.56,36.14,32.94,29.59,24.96,23.97,21.22,18.16,16.38.
因此,可确定上述化合物3m为3'-对溴苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例15:3'-间乙炔苯胺基-2'-羟基去氢枞酸丙酯(化合物3n)的制备
称量化合物2(120mg,0.34mmol)、间氨基苯乙炔(48.03mg,0.41mmol)和六合水高氯酸锌(5mg)溶于10ml无水乙醇置于圆底烧瓶中,在温度为80℃下、回流反应至完全(TLC跟踪检测,约1h),自然冷却至室温后,浓缩,加入5ml乙酸乙酯和10ml水萃取,有机层无水硫酸钠干燥过夜,浓缩,用硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到无色油状液体3n123.42mg,产率76.63%。1H NMR(600MHz,DMSO)δ7.16(d,J=7.9Hz,1H),7.03–6.95(m,2H),6.82(s,1H),6.66(s,1H),6.60(t,J=7.1Hz,2H),4.04(dd,J=15.1,8.0Hz,1H),3.98(dd,J=12.8,6.0Hz,2H),3.82(s,1H),3.10(s,1H),3.00(s,1H),2.76(s,4H),2.50(s,3H),2.29(d,J=12.6Hz,1H),2.11(t,J=11.0Hz,1H),1.73(d,J=8.7Hz,4H),1.64(s,2H),1.58(d,J=9.2Hz,1H),1.32(s,2H),1.21(s,3H),1.15(s,3H),1.14(s,3H),1.13(s,3H).13C NMR(151MHz,DMSO)δ179.07,147.84,146.79,145.92,134.57,129.33,127.05,124.22,124.10,122.94,122.04,116.29,114.33,84.16,68.80,66.78,62.62,48.02,46.59,45.06,38.02,37.05,36.89,33.53,30.16,25.25,24.04,21.93,18.62,16.66.
因此,可确定上述化合物3n为3'-间乙炔苯胺基-2'-羟基去氢枞酸丙酯,其结构式如下式所示:
实施例16:(3-苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4a)的制备
称量化合物3a(100mg,0.22mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.32mmol,6eq),冰浴,将三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷缓慢滴加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=2:1~4:1),得到白色固体4a 60.65mg,,产率57.96%。1H NMR(600MHz,DMSO)δ7.43(d,J=44.0Hz,2H),7.25(d,J=7.6Hz,2H),7.07(s,1H),7.02(s,1H),6.95(s,1H),6.76(s,1H),4.96(s,1H),4.35(s,1H),4.19(d,J=18.8Hz,2H),3.82(s,1H),2.81–2.65(m,3H),2.50(s,1H),2.19(s,1H),1.96(s,1H),1.75–1.55(m,3H),1.51(d,J=10.3Hz,2H),1.31(s,1H),1.16(s,3H),1.15(d,J=3.6Hz,3H),1.14(d,J=5.5Hz,3H),1.07(s,3H).13C NMR(151MHz,DMSO)δ177.30,154.12,146.37,145.03,138.14,133.96,128.81,126.50,124.15,123.79,123.48,117.83,70.22,65.00,47.31,46.34,44.90,37.46,36.56,36.30,36.09,32.93,29.68,29.43,25.06,24.01,21.21,18.06,16.27.
因此,可确定上述化合物4a为(3-苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例17:(3-邻甲苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4b)的制备
称量化合物3b(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.32mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=2:1~4:1),得到无色油状液体4b 62.34mg,,产率60.63%。1H NMR(600MHz,DMSO)δ7.22(d,J=14.3Hz,2H),7.14(d,J=28.7Hz,2H),7.01(s,1H),6.91(s,1H),6.80(s,1H),4.97(s,1H),4.34(s,1H),4.22(s,1H),4.00(s,1H),2.84–2.71(m,3H),2.48(s,1H),2.32(s,1H),2.10(d,J=5.9Hz,3H),1.97(s,1H),1.82–1.68(m,3H),1.60(d,J=9.7Hz,2H),1.31(d,J=12.8Hz,2H),1.23(d,J=2.8Hz,3H),1.16–1.14(m,3H),1.13(s,6H).13C NMR(151MHz,DMSO)δ177.88,155.61,147.00,145.67,136.58,136.13,134.66,131.44,128.29,127.09,124.66,71.44,65.13,60.31,49.03,47.82,45.47,38.21,37.09,33.46,30.05,25.57,24.44,21.78,21.31,18.61,17.89,16.89,14.64.
因此,可确定上述化合物4b为(3-邻甲苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例18:(3-间甲苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4c)的制备
称量化合物3c(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.32mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到淡黄色油状液体4c 58.56mg,,产率54.36%。1H NMR(600MHz,DMSO)δ7.33(s,1H),7.25(s,1H),7.10(d,J=25.9Hz,2H),6.95(t,J=9.0Hz,1H),6.85(s,1H),6.76(s,1H),4.94(s,1H),4.38(s,1H),4.18(d,J=13.1Hz,2H),3.82–3.73(m,1H),2.72(d,J=7.6Hz,3H),2.50(s,1H),2.25(d,J=11.3Hz,3H),1.98(s,1H),1.75–1.57(m,3H),1.52(d,J=10.4Hz,2H),1.27(d,J=25.5Hz,2H),1.16(d,J=3.7Hz,3H),1.15(s,3H),1.14(s,3H),1.07(s,3H).13C NMR(151MHz,DMSO)δ177.32,154.01,146.36,144.99,138.14,134.14,128.63,126.49,124.16,123.78,118.38,115.13,70.22,64.99,47.32,46.43,44.93,37.49,36.59,36.07,32.92,29.70,25.00,24.01,21.27,18.07,16.30.
因此,可确定上述化合物4c为(3-间甲苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例19:(3-对甲苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4d)的制备
称量化合物3d(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.32mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到白色固体4d59.32mg,,产率57.69%。1H NMR(600MHz,DMSO)δ7.30(d,J=8.5Hz,2H),7.11–7.06(m,3H),6.97(s,1H),6.74(s,1H),4.93(s,1H),4.39(s,1H),4.18(d,J=9.1Hz,2H),3.76(s,1H),2.78–2.68(m,3H),2.50(s,1H),2.25(s,3H),2.23(s,1H),1.97(s,1H),1.66(d,J=13.3Hz,2H),1.57(s,1H),1.50(d,J=12.1Hz,2H),1.25(s,1H),1.16(d,J=3.0Hz,3H),1.15(d,J=3.0Hz,3H),1.13(s,3H),1.07(s,3H).13C NMR(151MHz,DMSO)δ177.34,154.06,146.48,145.10,135.78,134.15,132.47,129.27,126.49,124.14,123.72,117.92,70.20,64.77,47.31,46.34,44.64,37.47,36.49,32.95,29.44,24.97,23.99,21.30,20.37,18.05,16.26.
因此,可确定上述化合物4d为(3-对甲苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例20:(3-(2-甲氧基苯基)-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4e)的制备
称量化合物3e(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.26mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到无色油状液体4e66.12mg,,产率62.27%。1H NMR(600MHz,DMSO)δ7.25(s,1H),7.18(s,1H),7.14(s,1H),7.08(s,1H),7.00(s,1H),6.82(s,1H),6.73(s,1H),4.94(s,1H),4.35(s,1H),4.24(s,1H),4.02(s,1H),3.74(s,1H),3.67(s,1H),2.83–2.73(m,3H),2.50(s,1H),2.31(s,1H),2.12(s,1H),1.98(s,1H),1.83–1.68(m,3H),1.63(d,J=12.6Hz,2H),1.33(d,J=14.9Hz,2H),1.24(s,3H),1.17(dd,J=6.7,5.1Hz,3H),1.16(d,J=2.7Hz,3H),13C NMR(151MHz,DMSO)δ177.37,170.40,155.72,154.77,146.50,145.20,134.14,128.78,128.27,126.59,125.79,123.92,120.48,112.48,70.93,64.59,59.82,55.66,48.04,47.38,44.96,37.69,36.61,32.96,29.74,25.11,23.98,20.82,18.14,16.35,14.15.
因此,可确定上述化合物4e为(3-邻甲氧基苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例21:(3-对甲氧基苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4f)的制备
称量化合物3f(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.26mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到白色固体4f75.42mg,产率71.03%。1H NMR(600MHz,DMSO)δ7.34(s,1H),7.29(s,1H),7.10(s,1H),6.96(s,1H),6.82–6.75(m,3H),4.92(s,1H),4.38(s,1H),4.18(d,J=9.2Hz,2H),3.70(s,3H),2.74(t,J=17.9Hz,3H),2.50(s,1H),2.20(s,1H),1.97(s,1H),1.76–1.54(m,3H),1.52(d,J=11.5Hz,2H),1.35–1.17(m,2H),1.15(dd,J=6.8,3.8Hz,9H),1.07(d,J=3.4Hz,3H).13CNMR(151MHz,DMSO)δ177.38,155.49,154.20,146.47,145.14,134.18,131.32,126.51,124.18,123.69,120.10,119.81,113.99,70.09,64.86,55.24,47.33,46.64,44.95,44.60,37.47,36.48,36.33,32.94,29.41,24.95,23.94,21.30,18.03,16.24.
因此,可确定上述化合物4f为(3-对甲氧基苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例22:(3-邻氟苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4g)的制备
称量化合物3g(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.26mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到无色油状液体4g61.83mg,产率59.64%。1H NMR(600MHz,DMSO)δ7.39(s,1H),7.26(s,1H),7.18(d,J=24.8Hz,2H),7.04(s,1H),7.00(s,1H),6.80(s,1H),5.01(s,1H),4.37(s,1H),4.26(s,1H),4.11(d,J=9.0Hz,1H),3.78(s,1H),2.78(d,J=6.6Hz,2H),2.50(s,1H),2.29(s,1H),2.08(s,1H),1.81–1.69(m,3H),1.62(d,J=15.1Hz,2H),1.32(d,J=37.5Hz,2H),1.22(s,3H),1.16(dd,J=6.9,4.0Hz,6H),1.13(s,3H).13C NMR(151MHz,DMSO)δ177.83,157.77,156.12,155.46,146.99,145.62,134.56,129.02,127.68,127.04,125.67,125.22,124.69,124.37,116.98,71.72,65.12,48.50,47.87,45.40,38.07,37.11,36.61,33.43,30.18,25.60,24.46,21.72,18.60,16.80.
因此,可确定上述化合物4g为(3-邻氟苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例23:(3-间氟苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4h)的制备
称量化合物3h(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.26mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到白色固体4h71.28mg,产率68.76%。1H NMR(600MHz,DMSO)δ7.46(s,1H),7.26(d,J=54.7Hz,2H),7.06(s,1H),6.95(s,1H),6.81(s,1H),6.74(s,1H),4.99(s,1H),4.33(s,1H),4.22(d,J=28.8Hz,2H),3.80(s,1H),2.81–2.64(m,3H),2.50(s,1H),2.19(s,1H),1.91(s,1H),1.72–1.50(m,5H),1.29(s,1H),1.18–1.16(m,3H),1.15(d,J=2.7Hz,3H),1.13(d,J=3.2Hz,3H),1.07(d,J=2.9Hz,3H).
13C NMR(151MHz,DMSO)δ177.23,163.01,161.51,153.98,146.29,144.96,139.86,133.85,130.46,126.46,123.78,113.24,109.80,104.77,70.46,64.81,47.33,46.40,44.89,37.42,36.56,36.29,36.09,32.95,29.70,25.09,24.00,21.21,18.08,16.26.
因此,可确定上述化合物4h为(3-间氟苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例24:(3-对氟苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4i)的制备
称量化合物3i(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.26mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到白色固体4i58.46mg,产率56.39%。1H NMR(600MHz,DMSO)δ7.46(s,1H),7.40(s,1H),7.07(d,J=8.2Hz,2H),7.01(s,1H),6.92(s,1H),6.75(s,1H),4.94(s,1H),4.37(s,1H),4.19(s,1H),3.77(s,1H),2.82–2.64(m,3H),2.48(s,1H),2.19(s,1H),1.95(s,1H),1.63(dd,J=58.3,29.2Hz,3H),1.51(s,2H),1.25(d,J=33.3Hz,2H),1.15(s,3H),1.14(d,J=4.2Hz,3H),1.13(s,3H),1.05(d,J=4.0Hz,3H).13C NMR(151MHz,DMSO)δ177.78,159.47,157.87,154.60,146.98,145.63,135.15,134.65,127.00,124.64,124.19,120.29,115.83,70.72,65.40,47.82,47.00,45.10,37.98,36.98,36.54,33.44,29.89,25.43,24.48,21.80,18.54,16.73.
因此,可确定上述化合物4i为(3-对氟苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例25:(3-间氯苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4j)的制备
称量化合物3j(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.26mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到无色油状液体4j71.35mg,产率66.61%。1H NMR(600MHz,DMSO)δ7.71(s,1H),7.33(d,J=32.3Hz,2H),7.08(d,J=21.5Hz,2H),6.95(s,1H),6.75(s,1H),4.99(s,1H),4.38(s,1H),4.25(s,1H),3.81(s,1H),2.82–2.61(m,3H),2.50(s,1H),2.22(s,1H),1.98(s,1H),1.74–1.59(m,3H),1.52(d,J=9.9Hz,2H),1.27(d,J=25.2Hz,2H),1.16(d,J=2.7Hz,3H),1.15(d,J=2.7Hz,3H),1.14(d,J=3.2Hz,3H),1.07(d,J=4.8Hz,3H).13C NMR(151MHz,DMSO)δ177.25,153.92,146.44,145.11,139.55,133.84,133.45,130.41,126.46,124.08,123.77,123.14,117.36,116.22,70.53,64.76,47.30,46.26,44.73,37.52,36.52,36.29,32.94,29.50,25.09,23.96,21.21,18.06,16.24.
因此,可确定上述化合物4j为(3-间氯苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例26:(3-对氯苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4k)的制备
称量化合物3k(100mg,0.21mmol)溶于5ml四氢呋喃,加入0.22ml 6M氢氧化钠溶液(1.26mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到白色固体4k72.56mg,产率67.74%。1H NMR(600MHz,DMSO)δ7.43(d,J=9.0Hz,2H),7.29(d,J=9.0Hz,2H),7.09(s,1H),6.98(s,1H),6.75(s,1H),4.97(s,1H),4.39(s,1H),4.20(s,1H),3.78(s,1H),2.80–2.66(m,3H),2.50(s,1H),2.20(s,1H),1.96(s,1H),1.70–1.56(m,3H),1.51(s,2H),1.24(d,J=14.5Hz,2H),1.17(d,J=3.8Hz,3H),1.16(d,J=3.8Hz,3H),1.13(s,3H),1.07(s,3H).13C NMR(151MHz,DMSO)δ177.77,154.45,146.97,145.63,137.66,134.64,129.17,127.72,126.98,124.63,124.18,119.80,70.86,65.35,47.82,46.74,45.07,37.95,36.96,36.84,33.45,29.88,25.43,24.47,21.79,18.53,16.73.
因此,可确定上述化合物4k为(3-对氯苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例27:[3-间溴苯基-噁唑烷酮-5-基]去氢枞酸甲酯(化合物4l)的制备
称量化合物3l(100mg,0.19mmol)溶于5ml四氢呋喃,加入0.19ml 6M氢氧化钠溶液(1.14mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到白色固体4l74.53mg,产率70.73%。1H NMR(600MHz,DMSO)δ7.86–7.73(m,1H),7.42–7.32(m,1H),7.29–7.20(m,1H),7.15(s,1H),7.05(s,1H),6.93(s,1H),6.73(s,1H),5.15–4.85(m,1H),4.36(s,1H),4.19(s,1H),3.79(s,1H),2.80–2.64(m,3H),2.48(s,1H),2.19(s,1H),1.97(s,1H),1.78–1.57(m,3H),1.50(d,J=12.5Hz,2H),1.26(d,J=25.0Hz,2H),1.15(d,J=6.9Hz,3H),1.13(dd,J=4.2,2.7Hz,3H),1.12(d,J=3.5Hz,3H),1.06(s,3H).13C NMR(151MHz,DMSO)δ177.71,154.40,146.77,145.61,140.32,134.57,131.16,126.98,126.50,124.58,124.27,122.32,120.62,117.13,70.93,65.24,47.83,46.80,45.40,37.90,36.59,33.43,30.00,25.44,24.46,21.70,18.55,16.77.
因此,可确定上述化合物4l为(3-间溴苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例28:(3-对溴苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4m)的制备
称量化合物3m((100mg,0.19mmol)溶于5ml四氢呋喃,加入0.19ml 6M氢氧化钠溶液(1.14mmol,6eq),冰浴,三光气(29.67mg,0.1mmol)溶于5ml二氯甲烷加入上述圆底烧瓶中,搅拌反应至完全(TLC跟踪检测,约2h),加入水10ml,混合物用二氯甲烷(3×15ml)萃取,合并有机层,无水硫酸镁干燥,用硅胶柱纯化(石油醚:乙酸乙酯=4:1),得到白色固体4m68.48mg,产率64.99%。1H NMR(600MHz,DMSO)δ7.41(d,J=9.1Hz,2H),7.34(d,J=9.0Hz,2H),7.03(s,1H),6.96(s,1H),6.77(s,1H),4.97(s,1H),4.33(s,1H),4.17(d,J=42.7Hz,2H),2.85–2.65(m,3H),2.50(d,J=1.0Hz,1H),2.16(s,1H),1.89(s,1H),1.78–1.58(m,3H),1.49(d,J=12.1Hz,2H),1.28(s,1H),1.24(s,1H),1.19–1.16(m,3H),1.15(d,J=8.0Hz,3H),1.12(s,3H),1.05(s,3H).13C NMR(151MHz,DMSO)δ177.23,154.04,146.34,144.99,137.43,133.87,131.54,126.39,124.09,119.58,115.43,70.29,65.08,47.34,46.22,44.96,37.37,36.51,35.95,32.90,31.18,29.70,25.00,23.97,21.16,18.02,16.22.
因此,可确定上述化合物4m为(3-对溴苯基-噁唑烷酮-5-基)去氢枞酸甲酯,其结构式如下式所示:
实施例29:(3-间乙炔苯基-噁唑烷酮-5-基)去氢枞酸甲酯(化合物4n)的制备
称量化
去氢枞酸噁唑烷酮衍生物及其制备方法和应用专利购买费用说明

Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



























动态评分
0.0