






专利摘要
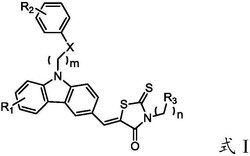
本发明公开了通式I的3‑甲基‑5,7‑二苯基‑5H‑噻唑并[3,2‑a]嘧啶‑2‑甲酰胺类衍生物或其药学可接受的水合物、盐,包括其立体异构体或互变异构体,其中,式I中的R1和R2分别独立的为氢、甲基、卤素、羟基、甲氧基、乙酰基、丙酰基、硝基或烷氧基;R3和R4独立的选自C1‑C6的烷基,或R3和R4与他们相连的氮原子一起组成吡咯基,哌啶基,吗啉基,N‑甲基哌嗪,N‑(4‑溴苯基哌嗪)。本发明的3‑甲基‑5,7‑二苯基‑5H‑噻唑并[3,2‑a]嘧啶‑2‑甲酰胺类衍生物作为血管内皮生长因子受体络氨酸激酶抑制剂,用于治疗和预防各种癌症的用途。
权利要求
1.一种VEGFR-2络氨酸激酶抑制剂,具有抗肿瘤活性,其特征在于:该化合物为具有通式I的3-甲基-5,7-二苯基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺类衍生物或其药学可接受的水合物、盐,包括其立体异构体或互变异构体;
其中,式I中的R1和R2分别独立的为氢、甲基、卤素、羟基、甲氧基、乙酰基、丙酰基、硝基或烷氧基;R3和R4独立的选自C1-C6的烷基,或R3和R4与他们相连的氮原子一起组成吡咯基,哌啶基,吗啉基,N-甲基哌嗪,N-(4-溴苯基哌嗪)。
2.根据权利要求1所述的化合物,其特征在于:所述R1为氟和氯。
3.根据权利要求1所述的化合物,包括:
5-(4-氯苯基)-7-(4-甲氧基苯基)-N,N-二甲基-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺;
5-(4-氯苯基)-7-(4-甲氧基苯基)-N,N-二乙基-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺;
5-(4-氯苯基)-7-(4-甲氧基苯基)-2- (哌啶-1-羰基)-3-甲基-5H-噻唑并[3,2-a]嘧啶;
5-(4-氯苯基)-7-(4-甲氧基苯基)-2-(吗啉-4-羰基)-3-甲基-5H-噻唑并[3,2-a]嘧啶;
5-(4-氯苯基)-7-(4-甲氧基苯基)-2-(4-甲基哌嗪-1-羰基)-3-甲基-5H-噻唑并[3,2-a]嘧啶;
5-(4-氯苯基)-7-(4-甲氧基苯基)-2-(吡咯烷-1-羰基)-3-甲基-5H-噻唑并[3,2-a]嘧啶;
5-(4-氯苯基)-7-(4-甲氧基苯基)- N,N-二正丁基-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺;
5-(4-氯苯基)-7-(4-甲氧基苯基)- 2-[4-(4-溴苯基)哌嗪-1-羰基]-3-甲基-5H-噻唑并[3,2-a]嘧啶。
4.权利要求1所述的化合物用于制备抗肿瘤药物的用途。
说明书
技术领域
本发明属于医药技术领域,尤其涉及3-甲基-5,7-二苯基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺类衍生物及其制备方法与作为血管内皮生长因子受体-2络氨酸激酶抑制剂,用于治疗和预防各种癌症的用途。
背景技术
恶性肿瘤是当今社会威胁人类健康的主要疾病之一,1971年,Folkman提出肿瘤的发生、发展与血管生成密切相关。肿瘤的生长和转移依赖于血管生成所提供的氧和必要的营养物质。实体肿瘤形成之初尚无血管生成,肿瘤细胞的营养主要通过弥散获得,当细胞距离毛细血管200 μm以上时,营养将无法通过弥散作用抵达肿瘤细胞,当肿瘤的细胞数达到1×107个以后,弥散获取的营养已无法满足肿瘤存活和生长的需要,此时如再无新的血管生成,肿瘤组织将发生退化。30多年来,大量的临床实践与实验研究证实:血管生成是肿瘤生长和转移的先决条件,也是所有恶性肿瘤的共性,抑制肿瘤介导的血管生成可以有效地抑制肿瘤的生长和转移。
血管生成是一个受众多调节、调控因子调节的复杂生理、病理过程,目前已分离和纯化出了20多种血管生成因子和相关因子,至少15种血管生成抑制剂。其中血管内皮细胞生长因子(vascular endothet ial growth factor,VEGF)处于核心地位,是目前已知活性最强、专属性最高的血管生成因子。血管内皮细胞生长因子受体(Vascular Endothelial Growth Gactor Receptor, VEGFR)是VEGF特异性的膜受体, 具有高亲和力。目前,一致的看法是VEGFR-2是VEGF的促有丝分裂、促血管生成和渗透性改变的主要中介因子。另外,VEGFR-2几乎只特定地在内皮细胞上表达,而且内皮细胞膜因稳定不易突变,通过抑制VEGFR-2可专属、有效地抑制肿瘤细胞血管生成,且不易产生耐药性。因此,以新生血管为靶点对肿瘤进行治疗也成为近年来的研究热点。
发明内容
本发明的目的在于提供一种3-甲基-5,7-二苯基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺类衍生物,其具有良好的抗肿瘤活性。
本发明的上述目的是通过如下技术方案来实现的:一种VEGFR-2络氨酸激酶抑制剂,具有抗肿瘤活性,其特征在于:该化合物为具有通式I的3-甲基-5,7-二苯基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺类衍生物或其药学可接受的水合物、盐,包括其立体异构体或互变异构体;
其中,式I中的R1和R2分别独立的为氢、甲基、卤素、羟基、甲氧基、乙酰基、丙酰基、硝基或烷氧基;R3和R4独立的选自C1-C6的烷基,或R3和R4与他们相连的氮原子一起组成吡咯基,哌啶基,吗啉基,N-甲基哌嗪,N-(4-溴苯基哌嗪)。
本发明中应用的术语“卤素”包括氟、氯或溴。
本发明还提供了上述化合物用于制备抗肿瘤药物的用途。
“药学上可接受的盐”指保留了式Ⅰ化合物的生物效力和性质,并与合适的非毒性有机或无机酸或有机或无机碱形成的常规酸加成盐或碱加成盐。酸加成盐的实例包括醋酸盐,己二酸盐,藻酸盐,天冬氨酸盐,苯甲酸盐,苯磺酸盐,硫酸氢盐,丁酸盐,柠檬酸盐,樟脑酸盐,樟脑磺酸盐,环戊丙酸盐,二葡萄糖酸盐,十二烷基硫酸盐,乙磺酸盐,富马酸盐,葡庚糖酸盐,甘油磷酸盐,半硫酸盐,庚酸盐,己酸盐,氢氯酸盐,氢溴酸盐,氢碘酸盐,2-羟基乙磺酸盐,乳酸盐,马来酸盐,甲磺酸盐,2-萘磺酸盐,烟酸盐,硝酸盐,草酸盐,扑酸盐,果胶酯酸盐,过硫酸盐,3-苯基丙酸盐,苦味酸盐,新戊酸盐,丙酸盐,琥珀酸盐,硫酸盐,酒石酸盐,硫氰酸盐,甲苯磺酸盐和十一酸盐。碱盐包括铵盐,碱金属盐,例如钠和钾盐,碱土金属盐,例如钙和镁盐,有机碱的盐,例如二环己胺盐,N-甲基-D-葡糖胺盐,和氨基酸的盐,例如精氨酸,赖氨酸等,而且,碱性含氮基团可以用这样的试剂季铵化,例如低级烷基卤化物,如甲基,乙基,丙基和丁基的氯,溴和碘化物;硫酸二烷基酯,如硫酸二甲酯,二乙酯,二丁酯和二戊酯;长链卤化物,如癸基,月桂基,肉豆蔻基和硬脂酰基的氯,溴和碘化物;芳烷基卤化物,如苄基和苯乙基的溴化物等。优选用于生成酸加成盐的酸包括盐酸和醋酸。
本发明还提供了上述通式I化合物的制备方法,该方法见下式。
本发明系统研究和阐释了所述化合物的结构和制备,所述化合物作为一类新的VEGFR-2络氨酸激酶抑制剂,结构类型新颖,为开发新型抗肿瘤药物提供了全新的方向和广阔的平台。
具体实施方式
实施例1
5-(4-氯苯基)-7-(4-甲氧基苯基)-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酸的制备
在250 mL的圆底烧瓶中加入4-氯苯甲醛0.1 mol、硫脲0.1 mol、4-甲氧基苯乙酮0.11 mol、三甲基氯硅烷0.1mol,乙腈30 mL,搅拌,回流反应10 h。冷却,抽滤,滤饼无水乙醇重结晶,得到黄色晶体4-(4-氯苯基)-6-(4-甲氧基苯基)-3,4-二氢嘧啶-2(1H)-硫酮,收率63%。ESI-MS (m/z):331.2 (M+H)+。
将0.05mol4-(4-氯苯基)-6-(4-甲氧基苯基)-3,4-二氢嘧啶-2(1H)-硫酮加入到10mL10%的KOH溶液中,然后滴加0.05mol2-氯乙酰乙酸乙酯,加热反应1h后,冰水稀释有大量晶体析出,干燥得2-(4-(4-氯苯基)-6-(4-甲氧基苯基)-1,2,3,4-四氢嘧啶-2-硫)-3-氧代丁酸乙酯,收率45%。
将0.02mol2-(4-(4-氯苯基)-6-(4-甲氧基苯基)-1,2,3,4-四氢嘧啶-2-硫)-3-氧代丁酸乙酯加入到120℃85%PPA中,反应1h,反应结束后冷却至室温,加入冰水,有大量固体析出,抽滤,无水乙醇重结晶得到5-(4-氯苯基-7-(4-甲氧基苯基)-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酸乙酯,收率43%。
将0.01mol5-(4-氯苯基-7-(4-甲氧基苯基)-3-甲基-5H-噻唑[并3,2-a]嘧啶-2-甲酸乙酯加入到2mol/L的NaOH中,再加入20mL甲醇,室温反应2h,减压除去甲醇,用浓盐酸调pH=2,有大量固体析出,抽滤,无水乙醇重结晶得浅黄色粉末5-(4-氯苯基)-7-(4-甲氧基苯基)-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酸,收率56% ,ESI-MS (m/z): 413.1(M+H)+。
实施例2
5-(4-氯苯基)-7-(4-甲氧基苯基)-N,N-二甲基-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺(L1)的制备
10mmol二甲胺盐酸盐,10mmol5-(4-氯苯基)-7-(4-甲氧基苯基)-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酸,20mL二氯甲烷加入到圆底烧瓶中,然后再加入12mmolEDCI,12mmolHOBt和三乙胺10mmol,室温反应12h后,反应液依次用5%HCl,5%NaHCO3,饱和NaCl溶液洗,干燥后柱层析分离得白色固体,收率36%;1H-NMR(300MHz,DMSO),δ(ppm):7.48 (2H,d,J = 8.7Hz), 7.40 (2H,d,J = 8.4Hz),7.24 (2H,d,J = 8.7Hz),6.84 (2H,d,J = 8.4Hz),6.73 (H,d),6.21 (H,d),3.83 (3H,s),3.13 (3H,s),3.02 (3H,s),2.31 (3H,s); ESI-MS (m/z):440.2 (M+H)+。
实施例3
5-(4-氯苯基)-7-(4-甲氧基苯基)-N,N-二乙基-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺(L2)的制备
将二甲胺盐酸盐用二乙胺替换,合成方法参照实施例2,收率39%;1H-NMR(300MHz,DMSO),δ(ppm):7.52 (2H,d,J = 8.4Hz),7.37 (2H,d,J = 8.4Hz), 7.24 (2H,d,J = 8.7Hz),6.84 (2H,d,J = 8.4Hz),6.72 (H,d),6.20 (H,d),3.84 (3H,s),3.48 (2H,m),3.32 (2H,m),2.30 (3H,s),1.19 (3H,t),1.07 (3H,t),ESI-MS (m/z):468.1 (M+H)+。
实施例4
5-(4-氯苯基)-7-(4-甲氧基苯基)-2- (1-哌啶-1-羰基)-3-甲基-5H-噻唑并[3,2-a]嘧啶(L3)的制备
将二甲胺盐酸盐用哌啶替换,合成方法参照实施例2,收率29%;1H-NMR(300MHz,DMSO),δ(ppm):7.52 (2H,d,J = 8.4Hz),7.32 (2H,d,J = 8.4Hz),7.24 (2H,d,J = 8.7Hz),6.83 (2H,d,J = 8.4Hz),6.74 (H,d),6.15 (H,d),3.84 (3H,s),3.48 (2H,m),3.40 (2H,m),2.30 (3H,s),1.61 (2H,m),1.54 (2H,m),1.45 (2H,m);ESI-MS (m/z):480.2 (M+H)+。
实施例5
5-(4-氯苯基)-7-(4-甲氧基苯基)-2-(1-吗啉-1-羰基)-3-甲基-5H-噻唑并[3,2-a]嘧啶(L4)的制备
将二甲胺盐酸盐用吗啉替换,合成方法参照实施例2,收率35%;1H-NMR(300MHz,DMSO),δ(ppm):7.49 (2H,d,J = 8.4Hz),7.28 (2H,d,J = 8.4Hz),7.14 (2H,d,J = 8.4Hz),6.87 (2H,d,J = 8.4Hz),6.72 (H,d),6.12 (H,d),3.85 (3H,s),3.75 (4H,br),3.68 (4H,q),2.34 (3H,s);ESI-MS (m/z): 480.1(M+H)+。
实施例6
5-(4-氯苯基)-7-(4-甲氧基苯基)-2-[1-(4-甲基哌嗪)-1-羰基]-3-甲基-5H-噻唑并[3,2-a]嘧啶(L5)的制备
将二甲胺盐酸盐用N-甲基哌嗪替换,合成方法参照实施例2,收率33%;1H-NMR(300MHz,DMSO),δ(ppm):7.50 (2H,d,J = 8.4Hz),7.32(2H,d,J = 8.4Hz),7.14 (2H,d,J = 8.4Hz),6.88 (2H,d,J = 8.4Hz),6.71 (H,d),6.02 (H,d),3.83 (3H,s),3.64 (4H,t),2.46 (4H,t),2.39 (3H,d),2.32 (3H,s);ESI-MS (m/z): 495.0(M+H)+。
实施例7
5-(4-氯苯基)-7-(4-甲氧基苯基)-2-(1-吡咯烷-1-羰基)-3-甲基-5H-噻唑并[3,2-a]嘧啶(L6)的制备
将二甲胺盐酸盐用四氢吡咯替换,合成方法参照实施例2,收率31%;1H-NMR(300MHz,DMSO),δ(ppm):7.52 (2H,d,J = 8.4Hz),7.32(2H,d,J = 8.4Hz),7.15 (2H,d,J = 8.4Hz),6.86(2H,d,J = 8.4Hz),6.71 (H,d),6.02 (H,d),3.83 (3H,s),3.48 (4H,m),2.36 (3H,s),1.87 (4H,m);ESI-MS (m/z): 466.0(M+H)+。
实施例8
5-(4-氯苯基)-7-(4-甲氧基苯基)- N,N-二正丁基-3-甲基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺(L7)的制备
将二甲胺盐酸盐用二正丁基替换,合成方法参照实施例2,收率52%;1H-NMR(300MHz,DMSO),δ(ppm):7.52 (2H,d,J = 9.0Hz),7.28 (2H,d,J = 9.0Hz),7.17 (2H,d,J = 9.0Hz),6.79 (2H,d,J = 9.0Hz),6.68 (H,d,6.02 (H,d),3.83 (3H,s),3.41 (4H,m),2.41 (3H,s),1.62 (4H,m),1.32 (4H,m),0.96 (6H,m);ESI-MS (m/z): 524.3 (M+H)+。
实施例9
5-(4-氯苯基)-7-(4-甲氧基苯基)- 2-[1-(4-(4-溴苯基)哌嗪)-1-羰基]-3-甲基-5H-噻唑并[3,2-a]嘧啶(L8)的制备
将二甲胺盐酸盐用1-(4-溴苯基)哌嗪替换,合成方法参照实施例2,收率37%;1H-NMR(300MHz,DMSO),δ(ppm):7.48 (2H,d,J = 8.4Hz),7.40 (2H,d,J = 8.7Hz),7.14 (2H,d,J = 8.4Hz),7.01 (2H,d,J = 8.7Hz),6.95 (2H,d,J = 8.4Hz),6.88 (2H,J = 8.4Hz),6.71 (H,d),6.02 (H,d),3.80 (3H,s),3.37 (4H,t),3.22 (4H,t),2.41 (3H,s);ESI-MS (m/z):635.0(M+H)+。
实施例10
MTT法抗肿瘤活性测定试验
1.材料的准备:
试剂:含10%胎牛血清的RPMl640培养液,0.25%胰蛋白酶消化液,PBS(0.01mol/L,pH7.4),DMSO(分析纯),MTT溶液;
细胞株:人宫颈癌Hela细胞株。
2.方法:
(1)接种细胞:将对数生长期的Hela细胞,用0.25%胰蛋白酶消化,用含10%胎小牛血清的1640培养基配成单个细胞悬液,以每孔104个细胞接种到96孔板,每孔体积100mL;
(2)培养细胞:将培养板放入CO2孵箱,在37℃,100%相对湿度,含5%CO2、95%空气的培养箱中培养24 h;
(3)配制5 mg/mLMTT溶液:称取250 mg MTT,放入小烧杯中,加50 mL PBS溶液,电磁搅拌30min,用0.22μm的微孔滤膜除菌,于4℃保存备用,l周内使用;
(4)给药:在无菌条件下,将目标化合物10-7mol/L的混悬液以100 μ L/孔加入到96孔培养板中,每种化合物设3个平行,每培养板上设3个空白对照孔。放置CO2孵箱中在同种条件下继续培养;
(5)呈色:培养24 h后,每孔加10μLMTT溶液,在培养4h,终止培养,小心吸弃孔内培养上清液,悬浮细胞离心后再吸弃孔内培养上清液,每孔加150μLDMSO,振荡10min,使甲臜充分溶解;
(6)比色:选择570 nm波长,在酶联免疫监测仪上测定各孔光吸收值(OD值);
按照以下公式计算肿瘤细胞生长抑制率:
细胞生长抑制率=(1-实验组OD值/对照组OD值)x100%。
3.结果:实验表明,本发明的上述化合物对肿瘤细胞均有抑制作用,见表1。
5,7-二苯基-5H-噻唑并[3,2-a]嘧啶-2-甲酰胺类衍生物及应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











![一种噻唑并[2,3-b]噁唑酮类化合物及其制备方法和用途](https://www.zhichawang.com/images/CN110511233A/CN110511233A.jpg)

![一种噻唑并[4,5-b]吡啶-6-羧酸及其化学合成方法](https://www.zhichawang.com/images/CN110590813A/CN110590813A.jpg)

动态评分
0.0