

IPC分类号 : C07K5/078,C07K5/097,A61P35/00,A61P31/18







专利摘要
本发明公开了一种1,2,4‑三氮唑偶联的二氢杨梅素衍生物或其药学可接受的水合物,包括其立体异构体或互变异构体。本发明的1,2,4‑三氮唑偶联的二氢杨梅素衍生物具有抗HIV‑1和抗癌作用,可用于治疗HIV‑1和癌药物中的用途。本发明公开了其制法。
权利要求
1.1,2,4-三氮唑偶联的二氢杨梅素衍生物,如下式所示:
所述化合物的具体为:
2.根据权利要求1所述的1,2,4-三氮唑偶联的二氢杨梅素衍生物,其制备方法包括以下步骤:
3.权利要求1所述的1,2,4-三氮唑偶联的二氢杨梅素衍生物,其在制备抗HIV-1和抗癌药物中的应用。
说明书
技术领域
本发明涉及1,2,4-三氮唑偶联的二氢杨梅素衍生物,及其在制药中的应用,属于医药技术领域。
背景技术
二氢杨梅素为多羟基双氢黄酮醇,属于黄酮类化合物,具有抗菌、抗氧化、抗癌、保肝、护肝等作用。但二氢杨梅素溶液易发生氧化,不能存放于金属容器中,稳定性较差,必须避光保存,在常温和冷水中溶解度低,不适宜做成注射剂,脂溶性也较差,在油脂体系中的抗氧化表现也不尽人意,限制了它的应用。为了提高其稳定性和溶解性,扩宽和加强其生理活性,对二氢杨梅素进行结构修饰已是必然。
研究表明,黄酮类化合物可以与HIV-1整合酶的核心区域中的关键氨基酸残基发生氢键、π-Cation作用,具有潜在的抗HIV-1活性。二氢杨梅素结构属于黄酮类化合物,对其改造,有望获得抗HIV-1药物。
1,2,4-三氮唑类药物在抗菌、抗HIV、抗肿瘤、治疗关节炎等方面有显著疗效。1,2,4-三氮唑作为一种重要的药效基团,易与生物大分子形成氢键,结合力较强,含该基团的药物的生物利用度和药效较好。研究表明,1,2,4-三氮唑结构片段已经通过Huisgen-Click反应被引入多肽、核苷和糖类药物分子中发挥了重要作用。
氨基酸是生命必需物质,具有良好的水溶性和亲和性。研究表明,癌细胞对氨基酸的需求量大于正常细胞,将其引入到药物分子中,有利于药物到达细胞内发挥作用,提高抗癌药效。
发明内容
本发明的目的在于提供一种1,2,4-三氮唑偶联的二氢杨梅素衍生物或其药学可接受的水合物、盐,包括其立体异构体和互变异构体,其具有抗HIV-1和抗癌的作用。
本发明的另一目的在于提供所述一种1,2,4-三氮唑偶联的二氢杨梅素衍生物或其药学可接受的水合物、盐,包括其立体异构体和互变异构体的制备方法。
本发明的再一目的在于提供所述1,2,4-三氮唑偶联的二氢杨梅素衍生物或其药学可接受的水合物、盐,包括其立体异构体和互变异构体的用途。
以下对本发明进行详细描述。
本发明提供的1,2,4-三氮唑偶联的二氢杨梅素衍生物或其药学可接受的水合物、盐,包括其立体异构体或互变异构体,如下式所示:
式中,R1各自独立为H,酰基,甲基;R2各自独立为H,烃基,芳基,芳杂基,苯并芳杂基;R3各自独立为OH,烷氧基,氨基酸(酯),多肽(酯);n为1-10的整数。
所述的1,2,4-三氮唑偶联的二氢杨梅素衍生物具体结构实例如下:
本发明还提供了所述化合物的制备方法:
式中,R1各自独立为H,酰基,甲基;R2各自独立为H,烃基,芳基,芳杂基,苯并芳杂基;R3各自独立为OH,烷氧基,氨基酸(酯),多肽(酯);n为1-10的整数。
所述药学上可接受的盐是各种有机酸、无机酸与所述二氢杨梅素硝酸酯衍生物形成的盐。例如:盐酸、硫酸、氢溴酸、氢氟酸、氢碘酸、硝酸、乙酸、草酸、柠檬酸、马来酸、富马酸、琥珀酸、苹果酸、甲磺酸、甲苯磺酸等。
本发明的1,2,4-三氮唑偶联的二氢杨梅素衍生物具有抗HIV-1和抗癌活性。
通过以下实施例进一步举例说明本发明,但应注意本发明的范围并不受这些实施例的任何限制。
具体实施方式
实施例1
中间体(I)的制备:
N2保护下,将320 mg (1.0 mmol)二氢杨梅素溶于5mLDMF中,加入5滴吡啶和112mg (1.1 mmol)Ac2O,室温反应10h,减压蒸除溶剂,柱层析纯化(V氯仿:V乙醇= 6:1,4:1,2:1梯度淋洗),得7-乙酰基二氢杨梅素。
N2保护下,将362 mg (1.0 mmol)7-乙酰基二氢杨梅素溶于5mLDMF中,加入625 mg(4.4 mmol)MeI 和607mg (4.4 mmol)K2CO3, 室温反应过夜,过滤,加入10mL浓度为30%的乙二胺甲醇溶液,40℃反应8h,减压蒸干,柱层析纯化(V氯仿:V乙醇= 6:1,4:1,2:1梯度淋洗),得3,3’,4’,5’-四甲氧基二氢杨梅素(中间体Ia),收率33%。
N2保护下,称取1.6g(5mmol)二氢杨梅素(含量98%)置于50mL的三角瓶中,然后依次加入15ml醋酸酐和1.2g硼酸,混匀,避光放置3天,倒入冰水中研磨,有黄色硼酸络合物析出,过滤,将所得黄色固体物加入40mL含有10%的HC1溶液,于60℃反应30min,除去硼酸,有黄白色固体生成,过滤,用水淋洗至中性,收集固体,干燥后即得粗品。用EtOH重结晶,得白色晶体,即二氢杨梅素四乙酰化产物(中间体Ib),产率89%。m.p.205-206℃。
实施例2
中间体IIa-e的制备
N2保护下,称取376mg(1.0mmol)中间体Ia,加入7mL丙酮、455mg(3.3 mmol)K2CO3,188mg(1.0mmol)1,2-二溴乙烷和128mg(1.0mmol)3-氨基-5-羧基-1,2,4-三唑,50℃反应6h,用稀醋酸调pH2.5,减压蒸干,加入10mL乙酸乙酯,搅拌,过滤,浓缩,硅胶柱层析纯化(V石油醚:V乙酸乙酯 = 4:1),得到中间体IIa,收率77%。
用216mg(1.0mmol)1,4-二溴丁烷代替188mg(1.0mmol)1,2-二溴乙烷,按上法制得中间体IIb,收率79%。
用244mg(1.0mmol)1,6-二溴己烷代替188mg(1.0mmol)1,2-二溴乙烷,按上法制得中间体IIc,收率80%。
用488mg(1.0mmol)中间体Ib代替376mg(1.0mmol)中间体Ia,按上法制得中间体IId,收率77%。
用202mg(1.0mmol)1,3-二溴丙烷代替188mg(1.0mmol)1,2-二溴乙烷,用488mg(1.0mmol)中间体Ib代替376mg(1.0mmol)中间体Ia,按上法制得中间体IIe,收率75%。
实施例3
化合物(1)的制备
称取167mg(1.2mmol)L-丙氨酸甲酯盐酸盐,加入10mLCH2Cl2和166mg(1.2 mmol)K2CO3,搅拌1h,再加入530mg(1.0mmol)中间体IIa、213mg(1.1 mmol)DCC和148mg (1.1mmol)HOBt,室温反应12h,过滤,浓缩,加15mL乙酸乙酯溶解,过滤,水洗涤(3x10mL),干燥,浓缩,硅胶柱层析纯化(V石油醚:V乙酸乙酯 = 5:1),得到化合物(1)。收率83%;ESI-MS (m/z): 615 [M]
实施例4
化合物(2)的制备
称取615mg(1.0mmol)化合物(1),加入甲醇7mL和1mol/L NaOH溶液7mL,室温搅拌1h,用醋酸调溶液pH2.5,减压蒸干,加入乙酸乙酯10mL,搅拌,过滤,水洗涤(3x10mL),干燥,浓缩,得到化合物(2),收率93%。
实施例5
化合物(3)的制备
用258mg(1.2mmol)D-苯丙氨酸甲酯盐酸盐代替167mg(1.2mmol)L-丙氨酸甲酯盐酸盐,其它操作同实施例3和4,得到化合物(3),总收率74%;ESI-MS (m/z): 677 [M]
实施例6
化合物(4)的制备
用200mg(1.2mmol)L-缬氨酸甲酯盐酸盐代替167mg(1.2mmol)L-丙氨酸甲酯盐酸盐,用438mg(1.0mmol)中间体IIb代替530mg(1.0mmol)中间体IIa,其它操作同实施例3和4,得到化合物(4),总收率76%;ESI-MS (m/z): 657 [M]
实施例7
化合物(5)的制备
用235mg(1.2mmol)L-Ala-Gly-OCH3盐酸盐代替167mg(1.2mmol)L-丙氨酸甲酯盐酸盐,其它操作同实施例3和4,得到化合物(5),总收率70%;ESI-MS (m/z): 658 [M]
实施例8
化合物(6)的制备
用305mg(1.2mmol)L-色氨酸甲酯盐酸盐代替167mg(1.2mmol)L-丙氨酸甲酯盐酸盐,用586mg(1.0mmol)中间体IIc代替530mg(1.0mmol)中间体IIa,其它操作同实施例3和4,得到化合物(6),总收率71%;ESI-MS (m/z): 772 [M]
实施例9
化合物(7)的制备
用642mg(1.0mmol)中间体IId代替530mg(1.0mmol)中间体IIa,其它操作同实施例3,得到化合物(7),收率80%;ESI-MS (m/z): 727 [M]
实施例10
化合物(8)的制备
用656mg(1.0mmol)中间体IIe代替530mg(1.0mmol)中间体IIa,用241mg(1.2mmol)D-苯甘氨酸甲酯盐酸盐代替167mg(1.2mmol)L-丙氨酸甲酯盐酸盐,其它操作同实施例3,得到化合物(8),总收率75%;ESI-MS (m/z): 789 [M]
实施例11
化合物(9)的制备
用320mg(1.0mmol)二氢杨梅素代替376mg(1.0mmol)中间体Ia,其它操作同实施例2和3,得到化合物(9),收率69%;ESI-MS (m/z): 559 [M]
实施例12
化合物(10)的制备
取559mg(1.0mmol)化合物(9),按实施例3操作,得到化合物(10),收率89%。
实施例13
将789mg(1mmol)化合物(8)溶于10mL乙酸乙酯,再加入5mL氯化氢-乙酸乙酯的饱和溶液,搅拌6h,过滤,干燥,得到化合物(8)的盐酸盐。
实施例14
1,2,4-三氮唑偶联的二氢杨梅素衍生物的抗HIV-1活性
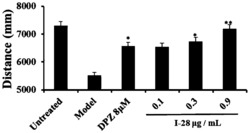
将测试的药物用0.01mol/L的PBS溶解,过滤,再用适量的DMEM无血清培养液倍比稀释至需要的浓度。采用MAGI-Assay检测药物的体外药效学效果。分别设立药物各项测试组,细胞对照组(CC),病毒对照组(VC),叠氮胸苷(AZT)阳性对照组,其中药物测试组在加毒后立即加入不同浓度的药物测试液,再按照MAGI-Assay操作规程进行活性测试,按如下公式计算抑制率(表1)。结果显示,1,2,4-三氮唑偶联的二氢杨梅素衍生物对HIV-1有好的抑制活性。
抑制率(%) = (病毒对照组蓝斑数-药物组蓝斑数)/病毒对照组蓝斑数 x 100%
实施例15
1,2,4-三氮唑偶联的二氢杨梅素衍生物对HepG2的抑制活性
取对数生长期的细胞,先用0.25%的胰酶消化3-5min,用含10%胎牛血清的DMEA培养液配成单个细胞悬液,以每孔2x10
1,2,4-三氮唑偶联的二氢杨梅素衍生物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0