

IPC分类号 : C07D213/12,C07D213/16,C07D213/26,C07D409/04,C07D409/14,C07D405/14








专利摘要
本发明提供了一种合成三取代吡啶衍生物的方法,属于吡啶衍生物的合成技术领域。一种合成三取代吡啶衍生物的方法,在三氟甲磺酸的存在下,由胺化合物和酮化合物反应合成吡啶衍生物,反应式如下:????????????????????????????????????????????????。本方法不仅能够适用于大量的官能团,而且操作简单、产率高,产物结构单一,便于分离和提纯、安全廉价、污染小。
权利要求
1.一种合成三取代吡啶衍生物的方法,在三氟甲磺酸的存在下,其特征在于:由式Ⅰ所示的胺化合物和式Ⅱ所示的酮化合物反应合成式Ⅲ所示的吡啶衍生物,
式Ⅰ: ;式Ⅱ: ;式Ⅲ: ;
其中,
R1选自苯基、甲苯基、噻吩基、苯甲基、C1-C7烷基中的一种,其中,R1取代基中的苯基、甲苯基、噻吩基、苯甲基、C1-C7烷基中的任何CH、CH2或CH3基团任选在所述CH、CH2或CH3基团上带有1、2或3个可相同或不同的以下的取代基:卤素或硝基;
R2选自苯基、甲苯基、噻吩基、苯并呋喃基、萘基中的一种,其中,R2取代基中的苯基、甲苯基、噻吩基、苯并呋喃基、萘基中的任何CH、CH2或CH3基团任选在所述CH、CH2或CH3基团上带有1、2或3个可相同或不同的以下的取代基:卤素或硝基;
卤素为氟、氯、溴或碘的取代基。
2.如权利要求1所述的合成三取代吡啶衍生物的方法,其特征在于:所述式Ⅰ所示的胺化合物为苄胺、对甲基苄胺、对氟苄胺、间甲基苄胺、间氟苄胺、2-噻吩甲胺、2-苯基乙胺或正己胺。
3.如权利要求1所述的合成三取代吡啶衍生物的方法,其特征在于:所述式Ⅱ所示的酮化合物为苯乙酮、对甲基苯乙酮、间甲基苯乙酮、2-乙酰基噻吩、2-乙酰基苯并呋喃、2-萘乙酮或对氯苯乙酮。
4.如权利要求1所述的合成三取代吡啶衍生物的方法,其特征在于:所述式Ⅲ所示的吡啶衍生物为2,6-二苯基-4-对甲基吡啶、4-(4-氟苯基)-2,6-二苯基吡啶、2,6-二苯基-4-间甲基吡啶、4-(3-氟苯基)-2,6-二苯基吡啶、2,6-苯基-4-(2-噻吩基)吡啶、4-苄基-2,6-二苯基吡啶、4-戊基-2,6-二苯基吡啶、4-戊基-2,6-二对甲苯基吡啶、4-戊基-2,6-二间甲苯基吡啶、4-苯基-2,6-二-(2-噻吩基)吡啶、2,6-二-(2-苯并呋喃)-4-苯基吡啶、2,6-二-(2-萘基)-4-对甲苯基吡啶、4-(4-氟苯基)-2,6-二-(2-萘基)吡啶、2,6-二-(4-氯苯基)-4-苯基吡啶、4-苯基-2,6-二对甲苯基吡啶、2,4,6-对甲苯基吡啶、4-(4-氟苯基)-2,6-二对甲苯基吡啶、2,4,6-三间甲基吡啶或4-(间氟苯基)-2,6-二间甲苯基吡啶。
5.如权利要求1所述的合成三取代吡啶衍生物的方法,其特征在于,所述合成三取代吡啶衍生物的方法,具体步骤如下:在反应容器中依次加入摩尔比为4:3的式Ⅰ所示的酮化合物和式Ⅱ所示的胺化合物,接着加入催化剂三氟甲磺酸,所述催化剂的加入量为0.05mmol/mmol酮化合物,在115~125℃油浴中反应18~24小时,冷却至室温,加水淬灭反应,萃取,减压浓缩,经柱层析纯化,得到三取代吡啶衍生物产品。
6.如权利要求5所述的合成三取代吡啶衍生物的方法,其特征在于,所述柱层析纯化的条件为:洗脱剂为石油醚和乙酸乙酯的混合物,石油醚和乙酸乙酯的体积比为50:1。
说明书
技术领域
本发明属于吡啶衍生物的合成技术领域,具体涉及一种合成三取代吡啶衍生物的方法。
背景技术
吡啶类衍生物是一种重要的精细化工中间体,广泛应用于农药、医药橡胶助剂、表面活性剂、粘结剂和日用化工领域,随着人类社会的不断进步以及应用研究不断深入,对吡啶衍生物的需求量也急剧增加。因此,研究其新的、简单利于工业化的合成方法也十分必要,也必将产生较好的经济效益。
目前吡啶衍生物的合成方法尽管很多,然而,这些方法主要有气相法和液相法,却存在很大的局限性。例如,气相法是采用醋酸酸化乙醛,使乙醛聚合为三聚乙醛,然后同醋酸经过高压泵与氨水混合,反应需要高温(220~280℃)、高压(10~20MPa)。如果采用三氧化二铝为催化剂,反应需要的温度更高,达到500℃。目前气相法采用的其它催化剂产率,底物的适应性,反应条件很难同时满足社会需求。液相法包括醛酮-烯腈法、苄胺路线法和环戊二烯路线合成法等。但是这些方法也存在很多缺点:反应条件苛刻,反应温度高,有的需要高温高压,分离困难,反应的底物限制性较强,因此,利用一种方法合成吡啶衍生物很有限。另外,利用金属催化过程中,催化剂的活性有限,这些缺点造成制备过程的操作难度增加,危害操作人员健康,环境污染严重。然而,现有合成吡啶衍生物的方法普遍存在:需要活泼的反应底物、反应收率低、反应时间较长、副产物多难处理及反应的形式过于单一(导致所合成的产物有很大的局限性)以及反应过程需要大量的溶剂或金属催化剂等缺点。鉴于此,研发新颖、绿色环保的吡啶衍生物的制备方法显得尤为重要。
发明内容
本发明所要解决的技术问题是,针对现有技术的不足,提供一种操作简单,产率高,产物单一,便于分离和提纯的利用胺、酮衍生物合成吡啶衍生物的方法。
为解决上述技术问题,本发明所采用的技术方案是:
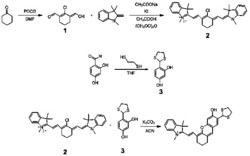
一种合成三取代吡啶衍生物的方法,在三氟甲磺酸(HOTf)的存在下,由式Ⅰ所示的胺化合物和式Ⅱ所示的酮化合物反应合成式Ⅲ所示的吡啶衍生物,
式Ⅰ: ;式Ⅱ: ;式Ⅲ: ;
其中,
R1选自苯基、甲苯基、噻吩基、苯甲基、C1-C7烷基中的一种,其中,R1取代基中的苯基、甲苯基、噻吩基、苯甲基、C1-C7烷基中的任何CH、CH2或CH3基团任选在所述CH、CH2或CH3基团上带有1、2或3个可相同或不同的以下的取代基:卤素或硝基;
R2选自苯基、甲苯基、噻吩基、苯并呋喃基、萘基中的一种,其中,R2取代基中的苯基、甲苯基、噻吩基、苯并呋喃基、萘基中的任何CH、CH2或CH3基团任选在所述CH、CH2或CH3基团上带有1、2或3个可相同或不同的以下的取代基:卤素或硝基;
卤素为氟、氯、溴或碘的取代基。
合成三取代吡啶衍生物的反应通式如下:
。
优选的,所述式Ⅰ所示的胺化合物为苄胺、对甲基苄胺、对氟苄胺、间甲基苄胺、间氟苄胺、2-噻吩甲胺、2-苯基乙胺或正己胺。
优选的,所述式Ⅱ所示的酮化合物为苯乙酮、对甲基苯乙酮、间甲基苯乙酮、2-乙酰基噻吩、2-乙酰基苯并呋喃、2-萘乙酮或对氯苯乙酮。
优选的,所述式Ⅲ所示的吡啶衍生物为2,6-二苯基-4-对甲基吡啶、4-(4-氟苯基)-2,6-二苯基吡啶、2,6-二苯基-4-间甲基吡啶、4-(3-氟苯基)-2,6-二苯基吡啶、2,6-苯基-4-(2-噻吩基)吡啶、4-苄基-2,6-二苯基吡啶、4-戊基-2,6-二苯基吡啶、4-戊基-2,6-二对甲苯基吡啶、4-戊基-2,6-二间甲苯基吡啶、4-苯基-2,6-二-(2-噻吩基)吡啶、2,6-二-(2-苯并呋喃)-4-苯基吡啶、2,6-二-(2-萘基)-4-对甲苯基吡啶、4-(4-氟苯基)-2,6-二-(2-萘基)吡啶、2,6-二-(4-氯苯基)-4-苯基吡啶、4-苯基-2,6-二对甲苯基吡啶、2,4,6-对甲苯基吡啶、4-(4-氟苯基)-2,6-二对甲苯基吡啶、2,4,6-三间甲基吡啶或4-(间氟苯基)-2,6-二间甲苯基吡啶。
优选的,所述合成三取代吡啶衍生物的方法,具体步骤如下:在反应容器中依次加入摩尔比为4:3的式Ⅰ所示的酮化合物和式Ⅱ所示的胺化合物,接着加入催化剂三氟甲磺酸,所述催化剂的加入量为0.05mmol/mmol酮化合物,在115~125℃油浴中反应18~24小时,冷却至室温,加水淬灭反应,萃取,减压浓缩,经柱层析纯化,得到三取代吡啶衍生物产品。
优选的,所述柱层析纯化的条件为:洗脱剂为石油醚和乙酸乙酯的混合物,石油醚和乙酸乙酯的体积比为50:1。
与现有技术相比,本发明的有益效果如下:本发明提供了一种利用胺、酮化合物在三氟甲磺酸的催化作用下合成吡啶衍生物的方法,该方法的反应底物易得,反应条件温和,无需高温高压处理,操作简便、安全,可高效地制备吡啶类化合物。本发明方法不仅能够适用于大量的官能团,而且操作简单、安全,产物的产率高、结构单一,便于分离和提纯,污染小。
具体实施方式
为了更好地理解本发明,下面结合实施例进一步清楚阐述本发明的内容,但本发明的保护内容不仅仅局限于下面的实施例。在下文的描述中,给出了大量具体的细节以便提供对本发明更为彻底的理解。然而,对于本领域技术人员来说显而易见的是,本发明可以无需一个或多个这些细节而得以实施。在其他的例子中,为了避免与本发明发生混淆,对于本领域公知的一些技术特征未进行描述。
下述实施例中,HOTf表示三氟甲磺酸,为其英文缩写。
实施例1
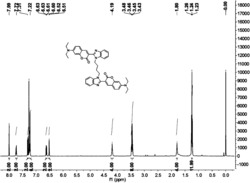
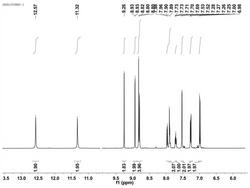
2,6-二苯基-4-对甲基吡啶的制备:在反应容器中加入苯乙酮1mmol(120mg),对甲基苄胺0.75mmol(91.6mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率92%,纯度为99.9%。1HNMR(400MHz,DMSO-d6)δppm:δ8.33(d,J=7.2Hz,4H),8.18(s,2H),7.96(d,J=8.4Hz,2H),7.49-7.58(m,6H),7.38(d,J=8.0Hz,2H);13CNMR(100MHz,DMSO-d6)δppm:δ156.4,149.9,139.5,139.3,135.2,130.2,129.7,129.2,127.6,127.4,116.7,21.3;HRMS(EI)Calcd.forC24H19N:[M+],321.1517.Found:m/z321.1521.
实施例2
4-(4-氟苯基)-2,6-二苯基吡啶的制备:在反应容器中加入苯乙酮1mmol(120mg),对氟苄胺0.75mmol(93.8mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率78%,纯度为99.8%1HNMR(400MHz,DMSO-d6)δppm:δ8.36(d,J=7.6Hz,4H),8.21(s,2H),8.15(q,2H),7.49-7.60(m,6H),7.43(t,2H);13CNMR(100MHz,DMSO-d6)δppm:δ164.7,162.2,157.0,148.9,139.2,134.6,130.1,129.7,129.2,127.4,116.5;HRMS(EI)Calcd.forC23H16NF:[M+],325.1267.Found:m/z325.1268.
实施例3
2,6-二苯基-4-间甲基吡啶的制备:在反应容器中加入苯乙酮1mmol(120mg),间甲基苄胺0.75mmol(91.6mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率91%,纯度为99.9%。1HNMR(400MHz,CDCl3)δppm:δ8.22(d,J=7.6Hz,4H),7.87(q,2H),7.41-7.55(m,9H),7.29(s,1H);13CNMR(100MHz,CDCl3)δppm:δ157.5,150.4,139.7,139.1,138.9,129.8,129.1,128.8,128.0,127.2,127.2,124.4,117.2,21.6;HRMS(EI)Calcd.forC24H19N:[M+],321.1517.Found:m/z321.1519.
实施例4
4-(3-氟苯基)-2,6-二苯基吡啶的制备:在反应容器中加入苯乙酮1mmol(120mg),间氟苄胺0.75mmol(93.8mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率87%,纯度为99.5%。1HNMR(400MHz,DMSO-d6)δppm:δ8.35(d,J=7.6Hz,4H),8.24(s,2H),7.91-8.00(m,2H),7.47-7.58(m,8H);13CNMR(100MHz,DMSO-d6)δppm:δ164.5,162.0,157.1,148.6,139.1,131.5,129.8,129.2,129.1,128.4,128.1,127.5,117.1;HRMS(EI)Calcd.forC23H16NF:[M+],325.1267.Found:m/z325.1270.
实施例5
2,6-苯基-4-(2-噻吩基)吡啶的制备:在反应容器中加入苯乙酮1mmol(120mg),2-噻吩甲胺0.75mmol(84.9mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率89%,纯度为99.7%。1HNMR(400MHz,DMSO-d6)δppm:δ8.29(d,J=7.2Hz,4H),8.12(s,2H),8.09-8.11(q,1H),7.91-8.00(m,2H),7.78–7.80(m,2H),7.48-7.58(m,6H),7.28-7.30(m,1H),13CNMR(100MHz,DMSO-d6)δppm:δ157.2,143.5,141.2,138.9,129.9,129.3,129.3,128.8,127.6,127.3,115.1;HRMS(EI)Calcd.forC21H15NS:[M+],313.0925.Found:m/z313.0926.
实施例6
4-苄基-2,6-二苯基吡啶的制备:在反应容器中加入苯乙酮1mmol(120mg),2-苯基乙胺0.75mmol(90.9mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率84%,纯度为99.6%。1HNMR(400MHz,CDCl3)δppm:δ8.09(d,J=7.2Hz,4H),7.42-7.48(m,6H),7.35-7.39(m,2H),7.19-7.31(m,5H),4.01(s,2H);13CNMR(100MHz,CDCl3)δppm:δ157.2,151.4,139.3,139.3,129.2,129.1,128.9,128.8,127.2,126.8,119.5,41.8;HRMS(EI)Calcd.forC24H19N:[M+],321.1517.Found:m/z321.1519.
实施例7
4-戊基-2,6-二苯基吡啶的制备:在反应容器中加入苯乙酮1mmol(120mg),正己胺0.75mmol(75.9mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率87%,纯度为99.7%。1HNMR(400MHz,CDCl3)δppm:δ8.14(d,J=7.2Hz,4H),7.39-7.51(m,8H),2.69-2.73(q,2H),1.68-1.73(m,2H),1.32-1.37(m,4H),0.89-0.92(q,3H);13CNMR(100MHz,CDCl3)δppm:δ156.9,153.2,139.8,128.8,128.7,127.1,119.1,35.8,31.5,30.3,22.6,14.1;HRMS(EI)Calcd.forC22H23N:[M+],301.1830.Found:m/z301.1833.
实施例8
4-戊基-2,6-二对甲苯基吡啶的制备:在反应容器中加入对甲基苯乙酮1mmol(134.2mg),正己胺0.75mmol(75.9mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率90%,纯度为99.9%。1HNMR(400MHz,CDCl3)δppm:δ7.96(d,J=8.0Hz,4H),7.78(s,2H),7.20(d,J=8.0Hz,4H),2.59-2.63(q,2H),2.33(s,6H),1.61-1.65(m,2H),1.28-1.29(m,4H),0.81-0.84(q,3H);13CNMR(100MHz,CDCl3)δppm:δ156.8,153.0,138.7,137.1,129.3,126.9,118.5,35.8,31.5,30.3,22.6,21.3,14.0;HRMS(EI)Calcd.forC24H27N:[M+],329.2143.Found:m/z329.2145.
实施例9
4-戊基-2,6-二间甲苯基吡啶的制备:在反应容器中加入间甲基苯乙酮1mmol(134.2mg),正己胺0.75mmol(75.9mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率88%,纯度为99.7%。(3j):1HNMR(400MHz,CDCl3)δppm:δ7.72-7.93(m,4H),7.39(s,2H),7.26-7.33(m,2H),7.13(d,J=6.8Hz,2H),2.59-2.63(q,2H),2.37(s,6H),1.61-1.64(m,2H),1.27-1.33(m,4H),0.81-0.85(q,3H);13CNMR(100MHz,CDCl3)δppm:δ157.1,153.1,139.9,138.2,129.6,128.6,127.8,124.3,119.2,35.8,31.6,30.4,22.6,21.7,14.1;HRMS(EI)Calcd.forC24H27N:[M+],329.2143.Found:m/z329.2144.
实施例10
4-苯基-2,6-二-(2-噻吩基)吡啶的制备:在反应容器中加入2-乙酰基噻吩1mmol(126.2mg),苄胺0.75mmol(80.4mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率77%,纯度为99.5%。1HNMR(400MHz,CDCl3)δppm:δ7.62-7.66(m,6H),7.38-7.49(m,5H),7.08-7.11(t,2H),;13CNMR(100MHz,CDCl3)δppm:152.7,150.2,144.9,138.6,129.2,129.2,128.0,127.9,127.1,124.9,115.1;HRMS(EI)Calcd.forC19H13NS2:[M+],319.0489.Found:m/z319.0484.
实施例11
2,6-二-(2-苯并呋喃)-4-苯基吡啶的制备:在反应容器中加入2-乙酰基苯并呋喃1mmol(160.2mg),苄胺0.75mmol(80.4mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率84%,纯度为99.6%。1HNMR(400MHz,CDCl3)δppm:δ8.08(s,2H),7.84(d,J=7.2Hz,2H),7.69(d,J=7.6Hz,2H),7.64(s,2H),7.48-7.60(m,5H),7.34(t,2H),7.29(t,2H);13CNMR(100MHz,CDCl3)δppm:δ155.4,155.2,150.1,149.9,138.1,129.4,129.2,128.9,127.2,125.3,123.3,121.8,116.8,111.6,105.5;HRMS(EI)Calcd.forC27H17NO2:[M+],387.1259.Found:m/z387.1265.
实施例12
2,6-二-(2-萘基)-4-对甲苯基吡啶的制备:在反应容器中加入2-萘乙酮1mmol(170mg),对甲基苄胺0.75mmol(91.6mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=40:1(v/v),得到白色固体产品,产率82%,纯度为99.9%。1HNMR(400MHz,CDCl3)δppm:δ8.64(s,2H),8.37(d,J=8.4Hz,2H),7.95-7.98(m,6H),7.87(d,J=8.4Hz,2H),7.66(d,J=7.6Hz,2H),7.50(m,4H),7.31(d,J=7.6Hz,2H),2.41(s,3H);13CNMR(100MHz,CDCl3)δppm:δ157.5,150.2,139.2,137.1,136.1,133.9,133.6,129.9,128.9,128.5,127.8,127.1,126.6,126.3,125.1,117.3,21.4;HRMS(EI)Calcd.forC32H23N:[M+],421.1830.Found:m/z421.1832.
实施例13
4-(4-氟苯基)-2,6-二-(2-萘基)吡啶的制备:在反应容器中加入2-萘乙酮1mmol(170mg),对氟苄胺0.75mmol(93.8mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=40:1(v/v),得到白色固体产品,产率78%,纯度为99.7%。1HNMR(400MHz,DMSO-d6)δppm:δ8.96(s,2H),8.59(dd,J=8.4,0.8Hz,2H),8.41(s,2H),8.20-8.22(m,2H),8.10-8.15(m,4H),8.00-8.02(m,2H),7.57-7.63(m,4H),7.44-7.57(t,2H);13CNMR(100MHz,DMSO-d6)δppm:δ150.7,149.1,136.6,134.6,133.9,133.6,130.2,130.1,129.2,128.7,128.1,127.3,127.0,126.8,125.3,116.4;HRMS(EI)Calcd.forC31H20NF:[M+],425.1580.Found:m/z425.1583.
实施例14:2,6-二-(4-氯苯基)-4-苯基吡啶的制备:在反应容器中加入对氯苯乙酮1mmol(154.6mg),苄胺0.75mmol(80.4mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率87%,纯度为99.7%。1HNMR(400MHz,DMSO-d6)δppm:δ8.16(d,J=7.6Hz,4H),7.90(s,2H),7.76(d,J=6.4Hz,2H),7.47-7.55(m,7H);13CNMR(100MHz,DMSO-d6)δppm:δ160.9,155.3,143.2,142.5,139.8,134.0,134.0,133.6,133.1,131.9,121.8;HRMS(EI)Calcd.forC23H15NCl2:[M+],375.0582.Found:m/z375.0786.
实施例15
4-苯基-2,6-二对甲苯基吡啶的制备:在反应容器中加入对甲基苯乙酮1mmol(134.2mg),苄胺0.75mmol(80.4mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率91%,纯度为99.9%。1HNMR(400MHz,CDCl3)δppm:δ8.08(d,J=8.0Hz,4H),7.81(s,2H),7.71(d,J=7.2Hz,2H),7.44-7.51(m,3H),7.29(d,J=8.0Hz,4H),2.40(s,6H);13CNMR(100MHz,CDCl3)δppm:δ152.7,145.3,134.6,134.2,132.2,124.7,124.4,124.1,122.5,122.3,111.8,16.6;HRMS(EI)Calcd.forC25H21N:[M+],335.1674.Found:m/z335.1679.
实施例16
2,4,6-对甲苯基吡啶的制备:在反应容器中加入对甲基苯乙酮1mmol(134.2mg),对甲基苄胺0.75mmol(91.6mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率95%,纯度为99.8%。1HNMR(400MHz,CDCl3)δppm:δ8.09(d,J=7.6Hz,4H),7.81(s,2H),7.63(d,J=7.2Hz,2H),7.30(d,J=6.4Hz,6H),2.42(s,6H);13CNMR(100MHz,CDCl3)δppm:δ157.4,149.9,138.9,137.0,129.8,129.4,127.0,116.3,21.3;HRMS(EI)Calcd.forC26H23N:[M+],349.1834.Found:m/z349.1830.
实施例17
4-(4-氟苯基)-2,6-二对甲苯基吡啶的制备:在反应容器中加入对甲基苯乙酮1mmol(134.2mg),对氟苄胺0.75mmol(93.8mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率87%,纯度为99.5%。1HNMR(400MHz,CDCl3)δppm:δ8.08(d,J=8.0Hz,4H),7.78(s,2H),7.69-7.73(t,2H),7.31(d,J=8.0Hz,2H),7.19-7.25(m,4H),2.43(s,6H);13CNMR(100MHz,CDCl3)δppm:δ164.6,162.1,157.5,149.0,139.1,136.8,129.4,129.0,128.9,127.0,116.0,21.4;HRMS(EI)Calcd.forC25H20NF:[M+],353.1580.Found:m/z353.1582.
实施例18
2,4,6-三间甲基吡啶的制备:在反应容器中加入间甲基苯乙酮1mmol(134.2mg),间甲基苄胺0.75mmol(91.6mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率89%,纯度为99.7%。1HNMR(400MHz,CDCl3)δppm:δ7.79–8.01(m,4H),7.85(s,2H),7.56(s,2H),7.39-7.43(m,3H),7.25-7.29(m,3H),2.48(s,9H);13CNMR(100MHz,CDCl3)δppm:δ157.7,150.2,139.7,139.1,138.8,138.3,129.8,129.7,129.0,128.6,127.9,127.9,124.4,117.3,21.7,21.6;HRMS(EI)Calcd.forC26H23N:[M+],349.1830.Found:m/z349.1833.
实施例19
4-(间氟苯基)-2,6-二间甲苯基吡啶的制备:在反应容器中加入间甲基苯乙酮1mmol(134.2mg),间氟苄胺0.75mmol(93.8mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率85%,纯度为99.9%。1HNMR(400MHz,CDCl3)δppm:δ7.97(m,4H),7.80(s,2H),7.38=7.52(m,5H),7.26(d,J=7.6Hz,2H),7.13-7.17(t,1H),2.47(s,6H);13CNMR(100MHz,CDCl3)δppm:δ164.5,162.1,157.9,148.8,139.4,138.4,130.7,130.0,128.7,127.9,124.4,122.9,116.6,21.7;HRMS(EI)Calcd.forC25H20NF:[M+],353.1580.Found:m/z353.1581.
实施例20
在反应容器中加入苯乙酮1mmol(120mg),苄胺0.75mmol(80.4mg),催化剂HOTf0.05mmol(7.5mg)。在120℃油浴中反应24小时,冷却至室温,加水淬灭反应,用乙酸乙酯洗涤三次,分液,合并有机层,活性炭脱色,过滤,无水硫酸钠干燥,减压浓缩,产品经过柱层析纯化,洗脱剂为石油醚:乙酸乙酯=50:1(v/v),得到白色固体产品,产率87%,纯度为99.7%。1HNMR(500MHz,CDCl3)ppm:8.43(d,J=8.0Hz,1H),8.34(d,J=8.0Hz,2H),8.00(d,J=8.5Hz,1H),7.92(s,1H),7.80(t,1H),7.51-7.64(m,9H);13CNMR(500MHz,CDCl3):156.90,149.23,149.05,139.77,138.56,130.35,129.70,129.60,129.50,128.96,128.72,128.52,127.75,126.47,125.92,125.75,119.39;HRMS(EI)Calcd.forC21H15N:[M+],281.1207;Found:281.1204。
一种合成三取代吡啶衍生物的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



![[1,5]苯并二氮卓类化合物的制备方法及其应用](https://www.zhichawang.com/images/10/CN104232073A/CN104232073A.jpg)

![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)








动态评分
0.0