









专利摘要
本发明公开了一种手性铂配合物及其制备方法,该配合物:二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ),具有手性。其以1,10-邻菲啰啉-5,6-二酮与2-羟基-4-甲氧基苯甲醛为原料,制备出2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉,再加入氯亚铂酸钾制备出2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·二氯合铂(Ⅱ),最后添加环己二胺,得到本发明的产品手性铂配合物。该配合物具有优异的抗肿瘤活性,能够应用于抗肝癌、肺癌和胃癌药物中。
权利要求
1.一种手性铂配合物,其特征在于:其结构式为:
。
2.根据权利要求1所述的手性铂配合物,其特征在于:该手性铂配合物化学名称为:二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)配合物。
3.据权利要求1或2所述的手性铂配合物,其特征在于:所述Pt是二价金属阳离子。
4.一种如权利要求1~3任一所述的手性铂配合物的制备方法,其特征在于:它的制备方法包括如下步骤:
(1) 按1,10-邻菲啰啉-5,6-二酮与2-羟基-4-甲氧基苯甲醛的摩尔比为1:1~ 2称取原料,按每摩尔的1,10-邻菲啰啉-5,6-二酮加入8~10 mL的冰醋酸,再按1,10-邻菲啰啉-5,6-二酮与乙酸铵的摩尔比为1: 25~30加入乙酸铵,在120~130℃油浴下,加热回流5~7 h,冷却至室温;在冰浴条件下,搅拌缓慢滴加浓氨水至中性,得到2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉的黄色沉淀物;
(2)再按每摩尔的2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉的黄色沉淀物中加入4~5 L的二甲基亚砜,油浴加热到140~150 ℃使黄色沉淀物溶解,得到黄色溶液;再将预先制备好的氯亚铂酸钾溶液缓慢滴入到黄色溶液中,析出黄色沉淀,继续回流搅拌1.5~2 h,使反应完全,趁热抽滤,除去未反应物,洗涤,过滤,烘干,得黄色的2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·二氯合铂(Ⅱ);
(3)每摩尔的二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉合铂(Ⅱ)中加入60~70 L的无水乙醇,70~80 ℃下反应20~30分钟后,加入2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·二氯合铂(Ⅱ)的6~8倍摩尔比的1, 2-环己二胺,恒温下回流至全溶解,过滤除去不溶物,滤液缓慢挥发,析出红色沉淀,用乙醇重结晶,即得到手性铂配合物。
5.根据权利要求4所述的手性铂配合物的制备方法,其特征在于:所述步骤(1)中浓氨水体积分数为25-28%。
6.根据权利要求4所述的手性铂配合物的制备方法,其特征在于:所述步骤(1)中浓氨水的加入量为每摩尔的1,10-邻菲啰啉-5,6-二酮滴加8~10 mL。
7.根据权利要求4所述的手性铂配合物的制备方法,其特征在于:所述步骤(2)中氯亚铂酸钾的制备为:先将氯亚铂酸钾加水,50~60 ℃下加热溶解,再按每摩尔的氯亚铂酸钾加0.8~1L的二甲亚砜,加热搅拌5 ~10min即可得到。
8.根据权利要求4所述的手性铂配合物的制备方法,其特征在于:所述步骤(2)中氯亚铂酸钾的加水量为每摩尔氯亚铂酸钾中加300~400 mL水。
9.根据权利要求1所述的手性铂配合物,其特征在于:它在制备抗肝癌、肺癌和胃癌药物中的应用。
说明书
技术领域
本发明涉及抗肿瘤分子化合物领域,具体涉及手性铂配合物及其制备方法和在制备抗肿瘤药物中的应用。
背景技术
自从人们发现G-四链体DNA是潜在的抗肿瘤靶标以来,人们在能稳定G-四链体DNA的有机小分子方面做了大量的研究工作。但是到目前为止绝大部分稳定G-四链体DNA的小分子为有机化合物,仅有一小部分是金属配合物。由于金属配合物具有很多有机小分子难以比拟的优点,如刚柔相济的几何结构变化和丰富的电化学性质,同时还具有光学、磁学以及催化等多种性能。在结合模式方面,金属配合物除了能与G-四链体DNA通过π-π堆积作用外,还能通过与碱基或者磷酸根骨架形成共价键的方式稳定G-四链体结构,进而达到抗肿瘤的效果。这均使得人们不断在金属配合物里寻找合适的抗肿瘤药物。
铂类配合物作为抗肿瘤药物的研究开始于二十世纪六十年代,在顺铂基础上铂类似物的设计合成与抗肿瘤活性筛选一直是抗肿瘤药物研究领域的热点。手性的铂类配合物其抗肿瘤活性也有高低之分,造成这种活性的差异与配合物结构的亲脂性有重要关系。
发明内容
本发明的目的就是提供具有抗肿瘤活性的手性铂配合物及其制备方法和应用。该配合物分子结构稳定,合成方法简单,同时具有很强的抗肿瘤活性,能为抗肿瘤药物的研发提供理论指导。
本发明的技术方案:
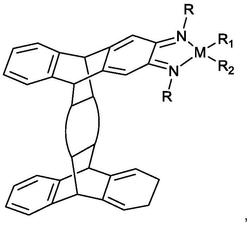
一种手性铂配合物,其结构式为:
该手性铂配合物化学名称为:二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)配合物,其具有手性,属于手性Δ型,Δ-[Pt(chda)(o-HO-p-MOPIP)]2+(Δ-Pt)。
本发明的手性铂配合物中Pt是二价金属阳离子。
本发明的手性铂配合物的制备方法,包括如下步骤:
(1)按1,10-邻菲啰啉-5,6-二酮(化合物1)与2-羟基-4-甲氧基苯甲醛的摩尔比为1:1~2称取原料,按每摩尔的1,10-邻菲啰啉-5,6-二酮加入8~10mL的冰醋酸,再按1,10-邻菲啰啉-5,6-二酮与乙酸铵的摩尔比为1:25~30加入乙酸铵,在120~130℃油浴下,加热回流5~7h,冷却至室温;在冰浴条件下,搅拌缓慢滴加浓氨水至中性,得到2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉的黄色沉淀物(化合物2);
(2)再按每摩尔的2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉的黄色沉淀物中加入4~5L的二甲基亚砜,油浴加热到140~150℃使黄色沉淀物溶解,得到黄色溶液;再将预先制备好的氯亚铂酸钾溶液缓慢滴入到黄色溶液中,析出黄色沉淀,继续回流搅拌1.5~2h,使反应完全,趁热抽滤,除去未反应物,洗涤,过滤,烘干,得黄色的2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·二氯合铂(Ⅱ)(配合物3);
(3)每摩尔的2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·二氯合铂(Ⅱ)(配合物3)中加入60~70L的无水乙醇,70~80℃下反应20~30分钟后,加入配合物3的6~8倍摩尔比的1,2-环己二胺(化合物4),恒温下回流至全溶解,过滤除去不溶物,滤液缓慢挥发,析出红色沉淀,用乙醇重结晶,即得到手性铂配合物(配合物5)。
作为技术方案的进一步优选,上述步骤(1)中浓氨水体积分数为25-28%,加入量为每摩尔的1,10-邻菲啰啉-5,6-二酮滴加8~10mL浓氨水。
作为技术方案的进一步优选,上述步骤(2)中氯亚铂酸钾的制备为:先将氯亚铂酸钾加水,50~60℃下加热溶解,再按每摩尔的氯亚铂酸钾加0.8~1L的二甲亚砜,加热搅拌5~10min即可得到。
作为技术方案的进一步优选,上述步骤(2)中氯亚铂酸钾的加水量为每摩尔氯亚铂酸钾中加300~400mL水。
本发明的化学反应式:
本发明各组分在手性铂配合物中的作用:
Pt++的作用:与DNA的某些亲核基团(如磷酸氧位点或碱基氮、氧位点)直接螯合,引起癌细胞的DNA损伤,使DNA在复制和转录当中受到障碍,从而阻止了癌细胞的生长和分裂,并导致其死亡。
二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)的作用:有利于插入DNA的双螺旋的碱基对之中。
本发明的手性铂配合物,应用到制备抗肝癌、肺癌和胃癌等抗肿瘤药物中的制备。
本发明的有益效果:
本发明的手性铂配合物分子结构稳定,合成方法简单。该配合物中咪唑并[4,5-f][1,10]-邻菲啰啉的引入,大大增强了配合物的细胞跨膜能力。同时抗肿瘤实验表明,该类配合物具有很强的抗肿瘤活性,其对NCI-H460细胞增殖的抑制能力为顺铂的10倍,在抗肿瘤药物方面将具有极大的应用潜力。
附图说明
图1为本发明的Δ-Pt手性铂配合物结构式;
图2为本发明Δ型手性铂配合物的红外光谱图,Δ-Pt的红外光谱为(KBr,cm–1):3303,2934,1613,1586,1457,1361,1327,1292,1208,1163,1082,1039,1026,809,717.配合物中的ν(C-N)在1292cm-1处,它在3303cm-1附近宽吸收峰可指认为ν(O-H)的伸缩振动。配合物中苯环的骨架伸缩振动位于1586cm-1。它在1208cm-1和1039cm-1处的吸收峰可分别指认为ν(C-O-C)的对称和不对称伸缩振动。
图3为本发明实施例1的产物对六种人肿瘤细胞BEL–7404(人肝癌细胞)、HepG2(人肝癌细胞)、NCI–H460(人肺癌细胞)、T–24(膀胱癌细胞)、MGC–803(人胃癌细胞)、A549(人肺腺癌细胞)和HL–7702(正常肝细胞)的抑制率图。
图4为本发明实施例1的产物对六种人肿瘤细胞BEL–7404(人肝癌细胞)、HepG2(人肝癌细胞)、NCI–H460(人肺癌细胞)、T–24(膀胱癌细胞)、MGC–803(人胃癌细胞)、A549(人肺腺癌细胞)和HL–7702(正常肝细胞)的IC50的柱状图。
图5为本发明实施例1的Δ-Pt手性铂配合物与端粒四链体Htel-1-DNA复合体系的紫外吸收光谱图(配合物的浓度为2.0×10–3mol/L,[DNA]/[配合物]=0–1)。
图6为本发明实施例1的Δ-Pt手性铂配合物与端粒四链体Htel-DNA复合体系的紫外吸收光谱图(配合物的浓度为2.0×10–3mol/L,[DNA]/[配合物]=0–1)。
图7为本发明实施例1的Δ-Pt手性铂配合物分别与端粒四链体DNA复合体系的荧光竞争图。
具体实施方式
下面通过实施例结合附图对本发明作进一步说明。
以下实施例中,化合物1:邻菲啰啉-5,6-二酮;
化合物2:2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉的黄色沉淀物;
配合物3:2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·二氯合铂(Ⅱ);
化合物4:1,2-环己二胺;
配合物5(手性铂配合物):本发明产品,二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)配合物。
实施例1
一、本发明的制备方法:
(1)化合物2的制备:称取2mol的邻菲啰啉-5,6-二酮(化合物1)与2mol的2-羟基-4-甲氧基苯甲醛,加入冰醋酸16mL和乙酸铵3.1g,在120℃油浴下,加热回流6h,冷却至室温;在冰浴条件下,搅拌,缓慢滴加16mL浓氨水(25-28%)至中性,得到2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉的黄色沉淀物(化合物2);
(2)配合物3的制备:预先制备氯亚铂酸钾溶液,在2.4mmol氯亚铂酸钾中,加水0.8mL,60℃加热溶解,加入2mL的二甲亚砜,60℃加热搅拌5min即可。
在2.4mmol化合物2中加入10mL的二甲基亚砜,油浴加热到140℃使化合物2溶解,将预先制备氯亚铂酸钾溶液缓慢滴入到化合物2溶液中,很快析出黄色沉淀,继续回流搅拌1.5h,使反应完全,趁热抽滤,除去未反应的配体和氯亚铂酸钾,用二甲亚砜、水、乙醇各洗涤一次,趁热过滤,烘干,得黄色的配合物3:2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·二氯合铂(Ⅱ)。
(3)手性铂配合物的制备:在0.6mmol配合物3中加入40mL的无水乙醇,70℃反应20分钟后,加入配合物3的8倍毫摩尔,即4.8mmol的环己二胺(化合物4),70℃回流至全溶解,过滤除去不溶物,滤液缓慢挥发,析出红色沉淀,用乙醇重结晶,得到手性铂配合物:二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)(配合物5)Δ-[Pt(chda)(o-HO-p-MOPIP)]2+,其结构式见图1。
二、产品检验:
产物经过电喷雾质谱、核磁共振谱、红外光谱和元素分析进行结构测定,确定目标配合物为Δ-[Pt(chda)(o-HO-p-MOPIP)]2+:C26H28PtN6O2,具体的波谱特性如下:
Δ-Pt核磁共振谱:1H NMR(400MHz,DMSO)δ8.97(d,1H),8.83(s,1H),8.20(d,1H),7.87(d,1H),7.31(s,1H),6.56–6.41(m,3H),3.95(s,1H),3.81(s,3H),2.15(d,2H),1.55(d,3H),1.20(s,2H).
Δ-Pt电喷雾质谱:m/z 651.02[M-2Cl-H]+。
元素分析C26H26PtN6O2实测值(计算值)/%:C 48.08(48.06);H 4.12(4.03);N 12.90(12.94);
因此,可确定上述红色沉淀为二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)配合物。
三、产品性能检测方法:
为了充分说明本发明所述的二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)配合物在制药中的用途,申请人以顺铂为阳性对照,对实施例1所得的配合物对多种人类肿瘤细胞株进行了体外抑制活性实验。
(1)用二甲基亚砜(DMSO)分别将Δ-Pt和顺铂配成2.0×10-3mol/L的储备液,缓冲溶液(pH=7.35)为0.1mol·L-1的三羟甲基氨基甲烷—盐酸(Tris-HCl),MTT试剂(3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐)的浓度为5mg/mL。
(2)BEL–7404(人肝癌细胞)、HepG2(人肝癌细胞)、NCI–H460(人肺癌细胞)、T–24(膀胱癌细胞)、MGC–803(人胃癌细胞)和A549(人肺腺癌细胞)细胞株均置于37℃、5%CO2充分湿化条件下的培养箱中,接种于含10%灭活新生牛血清的PPMI1640培养液中培养。
(3)将所有化合物配制成10μg/mL,助溶剂DMSO终浓度不超过1%,测试该浓度下各化合物对癌细胞的抑制率。
(4)取处于对数生长期的细胞,每孔180μL(约4500-5000个细胞)含细胞的培养基接种于96孔培养板,于37℃、5%CO2充分湿化条件下培养24h。
(5)待细胞贴壁后,按每孔20μL的量加入样品,每个样品设6个复孔,同时设定相应的空白对照。
(6)继续培养48h后,每孔加入10μL MTT试剂(浓度为5mg/mL),继续孵育4h后,吸弃上清液,每孔再加入150μL DMSO,轻微震荡反应5-8min,使结晶颗粒充分溶解。
(7)空白对照组调零,用酶标仪以490nm波长测定去除本底光吸收值后的吸光度值( 值),计算细胞增殖抑制率。可根据公式计算化合物的抑制率:抑制率=(1-加药组OD值/对照组OD值)×100%。
(8)所有实验均重复3次后取平均值。得到本发明的Δ-Pt和其对六种人肿瘤细胞的抑制率如图3和表1所示。
表1 MTT法分析配合物及顺铂对多种细胞的抑制率(%)
从图3及表1可见,本发明实施例1所制得的产物Δ-Pt,对六种人肿瘤细胞株均表现出显著的体外抗肿瘤活性,其抑制率分别在39.87至84.81之间。与临床用铂类药物顺铂相比,Δ-Pt对BEL–7404(人肝癌细胞)、NCI–H460(人肺癌细胞)和MGC–803(人胃癌细胞)三种细胞株,表现出比顺铂更高的体外增殖抑制活性。
实施例2
一、本发明的制备方法:
(1)化合物2的制备:称取2mol的邻菲啰啉-5,6-二酮(化合物1)与4mol的2-羟基-4-甲氧基苯甲醛,加入冰醋酸20mL和乙酸铵3.6g,在130℃油浴下,加热回流7h,冷却至室温;在冰浴条件下,搅拌,缓慢滴加20mL浓氨水(25-28%)至中性,得到黄色沉淀物(化合物2);
(2)配合物3的制备:预先制备氯亚铂酸钾溶液,在2.4mmol氯亚铂酸钾中,加水1mL,50℃加热溶解,加入3mL的二甲亚砜,50℃加热搅拌10min即可。
在2.4mmol化合物2中加入12mL的二甲基亚砜,油浴加热到150℃使化合物2溶解,将预先制备氯亚铂酸钾溶液缓慢滴入到化合物2溶液中,很快析出黄色沉淀,继续回流搅拌2h,使反应完全,趁热抽滤,除去未反应的配体和氯亚铂酸钾,用二甲亚砜、水、乙醇各洗涤一次,趁热过滤,烘干,得黄色的配合物3。
(3)手性铂配合物的制备:在0.6mmol配合物3中加入50mL的无水乙醇,80℃反应30分钟后,加入配合物3的7倍毫摩尔,即4.2mmol的(化合物4),80℃回流至全溶解,过滤除去不溶物,滤液缓慢挥发,析出红色沉淀,用乙醇重结晶,得到产品,手性铂配合物:二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)配合物(配合物5)Δ-[Pt(chda)(o-HO-p-MOPIP)]2+。
二、产品性能检测方法:
为了充分说明本发明所述的手性-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉-环己二胺铂配合物在制药中的用途,申请人以顺铂为阳性对照,对实施例1所得的配合物对多种人类肿瘤细胞株进行了体外抑制活性实验。
(1)用二甲基亚砜(DMSO)分别将Δ-Pt和顺铂配成2.0×10-3mol/L的储备液,缓冲溶液(pH=7.35)为0.1mol·L-1的三羟甲基氨基甲烷—盐酸(Tris-HCl),MTT试剂(3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐)的浓度为5mg/mL。
(2)BEL–7404(人肝癌细胞)、HepG2(人肝癌细胞)、NCI–H460(人肺癌细胞)、T–24(膀胱癌细胞)、MGC–803(人胃癌细胞)和A549(人肺腺癌细胞)细胞株均置于37℃、5%CO2充分湿化条件下的培养箱中,接种于含10%灭活新生牛血清的PPMI1640培养液中培养。
(3)将所有化合物配制成10μg/mL,助溶剂DMSO终浓度不超过1%,测试该浓度下各化合物对癌细胞的抑制率。
(4)取处于对数生长期的细胞,每孔180μL(约4500-5000个细胞)含细胞的培养基接种于96孔培养板,于37℃、5%CO2充分湿化条件下培养24h。
(5)待细胞贴壁后,按每孔20μL的量加入样品,每个样品设6个复孔,同时设定相应的空白对照。
(6)继续培养48h后,每孔加入10μL MTT试剂(浓度为5mg/mL),继续孵育4h后,吸弃上清液,每孔再加入150μL DMSO,轻微震荡反应5-8min,使结晶颗粒充分溶解。
所有实验均重复3次后取平均值。对初筛抗肿瘤效果好的受试化合物,继续用5个浓度梯度做相应细胞株的IC50值,得到本发明的Δ-Pt和其对六种人肿瘤细胞的IC50值如图4和表2所示。
表2 MTT法分析配合物及顺铂对六种人癌细胞株的细胞毒性
从体外增殖抑制活性测试结果来看,本发明实施例1所制得的产物Δ-Pt,对六种人肿瘤细胞株均表现出显著的体外抗肿瘤活性,其IC50值在1.91至19.59之间,与临床用铂类药物顺铂相比,Δ-Pt除对人肝癌细胞HepG2抗肿瘤活性较顺铂稍弱外,其对BEL–7404(人肝癌细胞)、NCI–H460(人肺癌细胞)、T–24(膀胱癌细胞)、MGC–803(人胃癌细胞)和A549(人肺腺癌细胞)五种细胞株,表现出比顺铂更高的体外增殖抑制活性,其中,Δ-Pt对NCI–H460(人肺癌细胞)的活性是顺铂的10倍。
以上数据表明,本发明所述的实施例1所制得的产物在抗肿瘤药物方面具有十分广阔的应用前景,有望用于制备肝癌、肺癌和胃癌的抗肿瘤药物。
实施例3
一、本发明的制备方法:
(1)化合物2的制备:称取2mol的邻菲啰啉-5,6-二酮(化合物1)与4mol的2-羟基-4-甲氧基苯甲醛,加入冰醋酸18mL和乙酸铵3.6g,在125℃油浴下,加热回流5h,冷却至室温;在冰浴条件下,搅拌,缓慢滴加18mL浓氨水(25-28%)至中性,得到黄色沉淀物(化合物2);
(2)配合物3的制备:预先制备氯亚铂酸钾溶液,在2.4mmol氯亚铂酸钾中,加水1mL,55℃加热溶解,加入2.5mL的二甲亚砜,55℃加热搅拌8min即可。
在2.4mmol化合物2中加入11mL的二甲基亚砜,油浴加热到145℃使化合物2溶解,将预先制备氯亚铂酸钾溶液缓慢滴入到化合物2溶液中,很快析出黄色沉淀,继续回流搅拌1.5h,使反应完全,趁热抽滤,除去未反应的配体和氯亚铂酸钾,用二甲亚砜、水、乙醇各洗涤一次,趁热过滤,烘干,得黄色的配合物3。
(3)手性铂配合物的制备:在0.6mmol配合物3中加入45mL的无水乙醇,75℃反应25分钟后,加入配合物3的6倍毫摩尔,即3.6mmol的环己二胺(化合物4),75℃回流至全溶解,过滤除去不溶物,滤液缓慢挥发,析出红色沉淀,用乙醇重结晶,得到产品,手性铂配合物:二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)配合物(配合物5)Δ-[Pt(chda)(o-HO-p-MOPIP)]2+。
二、产品性能检测方法:
(1)用10%二甲基亚砜(DMSO)将Λ-Pt和Δ-Pt配成2.0×10-3mol/L的溶液。
(2)Tris–HCl缓冲液:Tris 5mmol/L,用HCl滴定调节至pH=7.35,定容于1L容量瓶中,备用。
(3)用三羟甲基氨基甲烷—盐酸(Tris-HCl)缓冲溶液将端粒四链体DNA配成2.0×10-4mol/L的溶液。
(4)在比色皿中加入3mL,2.0×10-3mol/L的配合物溶液,逐次加入1μL,2.0×10-4mol/L的端粒四链体DNA溶液。
(5)每个混合液摇匀放置5min后,将其置于紫外-可见吸收光谱仪上扫描,结果如图5-6所示。
由图5-6可知,随着htel-1、htel浓度的增大,Δ-Pt-DNA复合体系的紫外吸收减少,其红移及减色率见表3。
Δ-Pt-DNA复合体系与DNA的键合强度Kb可以通过下列方程式确定:
[DNA]/(εa–εf)=[DNA]/(εb–εf)﹢1/[Kb(εb–εf)]
此处,εa,εf和εb分别是DNA的已知浓度,未与化合物键合及已与化合物键合的相关系数,Kb是化合物与DNA的键合常数,[DNA]是DNA在0.1mol/L缓冲溶液(pH=7.35)中的浓度。将[DNA]/(εa-εf)比[DNA]作图,可以得到斜率1/(εb–εf)和截距1/[Kb(εb–εf)],斜率和截距之比就可以得到键合常数Kb,Δ-Pt-DNA复合体系的键合常数见表2。由此可见,Δ-Pt-DNA较强地插入到DNA的碱基对之中,且还能通过沟槽键合与DNA作用。
由上述实施例可知,本发明的Δ-Pt具有稳定性好,与癌细胞DNA作用较强的特点,有望用于制备肝癌、肺癌和胃癌的抗肿瘤药物。
表3 化合物的红(蓝)移及减色率
实施例4
一、本发明的制备方法:
(1)化合物2的制备:称取2mol的邻菲啰啉-5,6-二酮(化合物1)与2mol的2-羟基-4-甲氧基苯甲醛,加入冰醋酸16mL和乙酸铵3.1g,在120℃油浴下,加热回流6h,冷却至室温;在冰浴条件下,搅拌,缓慢滴加16mL浓氨水(25-28%)至中性,得到黄色沉淀物(化合物2);
(2)配合物3的制备:预先制备氯亚铂酸钾溶液,在2.4mmol氯亚铂酸钾中,加水0.8mL,60℃加热溶解,加入2mL的二甲亚砜,60℃加热搅拌5min即可。
在2.4mmol化合物2中加入10mL的二甲基亚砜,油浴加热到140℃使化合物2溶解,将预先制备氯亚铂酸钾溶液缓慢滴入到化合物2溶液中,很快析出黄色沉淀,继续回流搅拌1.5h,使反应完全,趁热抽滤,除去未反应的配体和氯亚铂酸钾,用二甲亚砜、水、乙醇各洗涤一次,趁热过滤,烘干,得黄色的配合物3。
(3)手性铂配合物的制备:在0.6mmol配合物3中加入40mL的无水乙醇,70℃反应20分钟后,加入配合物3的8倍毫摩尔,即4.8mmol的环己二胺(化合物4),70℃回流至全溶解,过滤除去不溶物,滤液缓慢挥发,析出红色沉淀,用乙醇重结晶,得到手性铂配合物:二氯化-2-[2-(羟基)-4-(甲氧基)-苯基]咪唑并[4,5-f][1,10]-邻菲啰啉·S,S-环己二胺合铂(Ⅱ)配合物(配合物5)Δ-[Pt(chda)(o-HO-p-MOPIP)]2+。
二、产品性能检测方法:
(1)Tris–HCl缓冲液:Tris 5mmol/L,用HCl滴定调节至pH=7.35,定容于1L容量瓶中,备用。
(2)用10%二甲基亚砜(DMSO)将Λ-Pt和Δ-Pt配成2.0×10-3mol/L的溶液。
(3)将噻唑橙[TO]配成1.0×10-3的溶液,将CT-DNA、ds-26、Htel和Htel-1等四种G4-DNA配成1.0×10-4的溶液。
(4)在比色皿中加入1.0×10-3mol/L的噻唑橙(TO)3.0ul,1.0×10-4的G4-DNA 7.5ul,2990ul的三羟甲基氨基甲烷—盐酸(Tris-HCl)缓冲溶液(pH=7.35),逐次加入3uL的配合物溶液,使配合物对DNA的浓度逐渐提高。
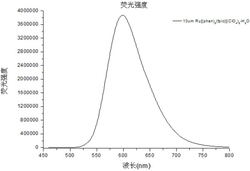
(5)将上述每个混合液用二次亚沸蒸馏水稀释至5mL后摇匀。摇匀放置5min后,将其置于荧光光谱仪上扫描(λex均为501nm,CT-DNA和ds-26的λem为526nm,Htel和Htel-1的λem为533nm),结果如图7所示。
如图7可见,配合物Δ-Pt与不同G–四链体DNA有很强的结合作用,Δ-Pt的DC50值在0.3–0.6μmol/L之间,与双螺旋DNA(ds26)作用的DC50值为1.0μmol/L。配合物与G–四链体DNA结合能力强于双螺旋DNA。
一种手性铂配合物及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0