

IPC分类号 : C07F5/00I,C09K11/06I,G01N21/33I,G01N21/64I








专利摘要
本发明公开了一种基于吡啶配体的双核镝配合物及其制备方法和应用。该配合物的化学式为[Dy2(L)2(NO3)4],L为2‑氨基‑1,2‑二‑吡啶‑乙醇脱去羟基氢原子,带一个单位负电荷;该配合物属于单斜晶系,I2/a空间群。本发明所述双核镝配合物的制备方法为:取六水合硝酸镝和2‑(氨甲基)‑吡啶置于混合溶剂中,在4‑甲氧基‑3‑羟基苯甲醛存在的条件下加热反应,反应物冷却,有晶体析出,收集晶体,即得。本发明所述的双核镝配合物对有机溶剂具有良好的发光响应,特别是对乙醇和乙腈具有良好发光响应,可以用于检测或识别不同的有机溶剂,因此,可用作敏化剂。
权利要求
1.基于吡啶配体的双核镝配合物,其特征在于:
该配合物的化学式为:[Dy2(L)2(NO3)4],其中,L为2-氨基-1,2-二-吡啶-乙醇脱去羟基氢原子,带一个单位负电荷;
该配合物属于单斜晶系,I2/a空间群,晶胞参数为
2.权利要求1所述基于吡啶配体的双核镝配合物的制备方法,其特征在于:取六水合硝酸镝和2-(氨甲基)-吡啶置于混合溶剂中,在4-甲氧基-3-羟基苯甲醛存在的条件下加热反应,反应物冷却,有晶体析出,收集晶体,即为目标配合物;其中,所述的混合溶剂为甲醇和乙腈的组合物。
3.根据权利要求2所述的制备方法,其特征在于:所述混合溶剂的组成中,甲醇和乙腈的体积比为2:1-1:2。
4.根据权利要求2所述的制备方法,其特征在于:反应在≥50℃的条件下进行。
5.根据权利要求2所述的制备方法,其特征在于:反应在60-120℃的条件下进行。
6.权利要求1所述的基于吡啶配体的双核镝配合物在制备乙醇和乙腈的敏化剂中的应用。
说明书
技术领域
本发明涉及稀土配合物,具体涉及一种基于吡啶配体的双核镝配合物及其制备方法和应用。
背景技术
配位化学在金属离子的光致发光探针的设计中起着至关重要的作用。金属与有机染料的配位引起了不同的光学反应,这表明它们之间存在能量转移。d6、d8和d10构型的发光镧系元素和过渡金属配合物往往表现出与有机染料不同的独特发光特性,如量子产率高、燃烧量大、发射波长和发射寿命长,由于对微环境的灵敏度低,可以作为探索发光物质金属离子,阴离子和中性物质的探针。在文献中,有详细讨论基于pct、esprit、fit和exciter形成机制的金属问题的设计原理和配位化学相关的研究。基于Ln3+和d6、d8以及d10金属配合物的荧光探针设计也通过讨论影响这些金属配合物荧光的某些因素得到了研究。对用于鉴定人体必需金属阳离子或环境中有毒金属阳离子的光致发光探针的研究发展迅猛,其重点是介绍其设计原理和传感行为。此外,还有一部分是结合金属配位相关的传感行为和设计方法,如讨论生物相关粒子ppi、中性生物分子三磷酸腺苷、一氧化氮和硫化氢的金属络合基光致发光探针。。
基于吡啶的配体,例如2,2'-bipyridine和1,10-phenanthroline,在超分子化学领域以及材料科学引起了人们的极大兴趣。在过去的几十年中,含有磷光的聚合物重d6过渡金属离子络合物吸引了很多注意,特别是因为这些金属配合物的实用性,如Ru(II),Ir(III),Os(II)和Re(I)离子在材料科学中的重要应用。通过将金属配合物嵌入聚合物结构中,所得材料的光物理和电化学性质可以通过其性质来调控金属配合物以及聚合物的属性:例如可加工性和成膜能力。同时,金属聚合物受到不同的科学研究领域的广泛关注:超分子化学,基础电子和能量转移,金属离子传感器以及电子设备。有些团队还广泛研究了含有过渡金属元素的金属聚合物的光伏和光功率限制应用。d6配置金属离子[即Ru(II),Ir(III),Os(II)和Re(I)]是所选择的过渡金属离子,由于它们特有的光谱性质,具备感光性和在可见光区域的广泛吸收波长。它们的化学稳定性,氧化还原性质,发光,激发态寿命和激发态反应性的独特相干性是众多衍生物被合成的触发因素。具有金属-配体电荷转移(MLCT)特征的晚期d6金属配合物的相对长寿命激发态促进了全面的光物理和光化学研究。
光致发光技术具备较高的灵敏度和选择性,较高的时间和空间分辨率,被认为是检测各种分析材料成分的有效手段之一。精细的仪器操作能力、商用可用性、较低的检测极限、原位和体内检测能力使该技术具有极大的吸引力。具有生物和环境意义的化学物种的光敏中心传感和成像已成为一个重要和快速发展的领域,这在很大程度上受到共聚焦显微镜和光学成像技术改进的刺激。近十年来,有大量具有各种特性的光致发光现象被报道出来。它们可以用于检测或成像特定分子、微环境、生物过程和事件。
在光致发光传感和探测中,金属配位诱导的光物理、电物理或生物性质的改变是必不可少的相关应用。大多数金属离子荧光探针是基于金属配位诱导的有机染料发射强度、寿命或波长的变化而研制的。金属配合物的荧光辐射来自有机染料π-π*激发态的辐射固定。基于d6、d8和d10构型的镧系Ln和过渡金属阳离子的光致发光探针不同于有机染料,后者的发光发射来自金属中心(MC,通过LMCT过程)或MLCT激发态的热驰豫。
将有机荧光团与特定的螯合剂(受体)结合作为荧光信号传送器是设计金属离子荧光探针的常用方法。在这种情况下,金属对受体的配位作用是关键。改变荧光强度、寿命或激发/发射最大值,检测金属阳离子的存在。荧光团和受体之间的分子内相互作用对于这些荧光探针的设计至关重要[9]。常规的PET机制,如光诱导电子转移光诱导电荷转移(PCT)的荧光共振能量转移。FRET和光诱导激子/激子丛的形成是探针分子构建的常用方法。另一方面,金属离子配位抑制激发态分子内质子转移(ESIPT)和聚集诱导等新理论也得到了广泛应用,发射(AIE)也被用来设计探针。
发射金属配合物具有量子产率高、燃烧位移大、发射波长长、寿命长、水溶性好等特点。它们的发射通常是不依赖于酸碱度的,对微环境的敏感度较低,与有机荧光团相比具有重要的优势。这些金属配合物是设计光致发光探针的潜在荧光报告器。迄今为止报道的最有趣的例子是基于发光的镧系配合物(Ln=Sm3+,Eu3+,Tb3+,Dy3+,Yb3+),或具有d6,d8和d10构型的过渡金属配合物。根据它们与系统间交叉(Wisc)有关的机制,它们的发射通常被称为磷光。
镧系化合物的磷光(Ln=Sm3+,Eu3+,Tb3+,Dy3+,Yb3+)来自激发诱导的f-f跃迁,f和f14构型的镧系元素中心的配合物是不发光的。对于半f填充的Ln3+,它的结构将导致它的基态和激发态之间的巨大能量差距,这使得它们很难在可见光窗口中显示发光。因为Ln3+离子的f-f跃迁是禁止的,则低摩尔消光系数不足以直接激发这些金属中心。这个问题可以通过在某些情况下结合电子来解决,它也可以作为Waco-Gilligan进入Ln3+复合物。当电子从基态到其单态的激发态被激发后,它就处于三线态。然后,分子内的能量从电子的三重态转移到Ln3+的激发态,然后是发光的f-f过渡到Ln的基态,在可见的范围内发射。由于发光机制,Ln配合物的发射寿命在毫秒和微秒之间,其发射现象也称为磷光或荧光。
与大多数有机染料(以纳秒为尺度)的短发射寿命相比,长发射寿命的Ln离子有利于通过时间门控检测细胞、组织等复杂微环境中的分析物来最小化光散射或自荧光的干扰。Ln3+络合物的发射光谱通常分离得很好。例如,Tb3+的主要f5、d4和f4-d4发射峰不与任何EUT发射峰重叠。此外,Ln3+的内壳轨道几乎不受其他因素的影响。因此,Ln3+配合物的发射带通常较窄(~20nm),为这些配合物提供了额外的优势。在多色成像中。反斯托克斯效应从天线吸收转变为LNT发射,可调量子产额是Ln复合物的附加光学特性,为发光探针的设计提供了一个适合的平台。Eu3+和Tb3+的发光配合物是最频繁的,因为它们的激发物对能量转移到O-H、N-H或C-H振荡器引起的振动猝灭效应的灵敏度较低,而这些振动猝灭效应经常出现在溶液和成像微环境中。
稀土配合物的量子产率有几个主要因素。电子三重态的能级应至少比Ln的激发态高1700cm,而过高或过低的能级只会导致天线的荧光消失。如果天线三重态的能级太接近Ln3+的d激发态,就会发生从Ln3+到天线的反向能量转移,从而导致Ln3+发光的降低[23]。另一方面,天线与Ln3+中心的距离有利于实现天线到LNT的能量传递。此外,Ln3+中心的协调环境也与其发光效率和寿命密切相关。由于Ln3+中心倾向于高配位数(>6),并显示出对硬配体的高亲和力,许多Ln3+配合物与水等溶剂进行配位交换,从而触发非发射散射过程,并显著降低量子产率。因此,天线配体和其他共配体的精细设计对于实现特定分析物的LNT基发光材料具有重要意义。目前还未见有基于吡啶配体的金属镝配合物的构筑及其荧光性质的相关报道。
发明内容
本发明要解决的技术问题是提供一种结构新颖且对乙酸和乙腈具有良好发光响应的基于吡啶配体的双核镝配合物及其制备方法和应用。
本发明所述的基于吡啶配体的双核镝配合物,其配合物的化学式为:[Dy2(L)2(NO3)4],其中,L为2-氨基-1,2-二-吡啶-乙醇脱去羟基氢原子,带一个单位负电荷;该配合物属于单斜晶系,I2/a空间群,晶胞参数为 α=90.00°,β=110.003(3)°,γ=90.00°。
本发明还提供上述基于吡啶配体的双核镝配合物的制备方法,具体为:取六水合硝酸镝和2-(氨甲基)-吡啶置于混合溶剂中,在4-甲氧基-3-羟基苯甲醛存在的条件下加热反应,反应物冷却,有晶体析出,收集晶体,即为目标配合物;其中,所述的混合溶剂为甲醇和乙腈的组合物。
上述制备方法中,六水合硝酸镝和2-(氨甲基)-吡啶的摩尔比为化学计量比,在实际操作过程中,六水合硝酸镝的量可相对过量一些。在混合溶剂的组成中,甲醇和乙腈的体积比为2:1-1:2。所述混合溶剂的用量可根据需要确定,通常以能溶解参加反应的原料为宜。具体地,以1mmol的2-(氨甲基)-吡啶为基准计算,全部原料所用混合溶剂的总用量通常为6-15mL。在具体溶解的步骤中,可将各原料分别用混合溶剂中的某一种组分溶解,然后再混合在一起反应;也可将所有原料混合在一起后再加混合溶剂溶解。
上述制备方法中,对于4-甲氧基-3-羟基苯甲醛,它的存在并未参与配位,但当它不存在时又无法得到本发明所述的目标配合物,因此,本申请人推测其起到催化的作用。所述4-甲氧基-3-羟基苯甲醛的用量通常为2-(氨甲基)-吡啶物质的量的0.1倍以上,优选为与2-(氨甲基)-吡啶物质的量相当。
上述制备方法中,反应优选是在≥50℃的条件下进行,进一步优选在60-120℃的条件下进行。当反应在60-120℃条件下进行时,反应时间通常控制在30-60h。
申请人研究发现,本发明所述的基于吡啶配体的双核镝配合物在不同的有机溶剂中的发光强度不同,可以用于检测或识别不同的有机溶剂。因此,本发明还包括上述基于吡啶配体的双核镝配合物在制备敏化剂中的应用。
与现有技术相比,本发明提供了一种结构新颖的基于吡啶配体的双核镝配合物及其制备方法,申请人在实验中发现,该双核镝配合物对有机溶剂具有良好的发光响应,特别是对乙酸和乙腈具有良好发光响应,可以用于检测或识别不同的有机溶剂,因此,可用作敏化剂。
附图说明
图1为本发明实施例1中反应完成后,冷却后的内衬管及其中的反应物的照片;其中,(a)为反应完成后冷却后的内衬管及其中的反应物的照片,(b)为内衬管底部晶体的放大照片。
图2为本发明实施例1制得的最终产物的红外光谱谱图。
图3为本发明实施例1制得的最终产物的粉末衍射谱图。
图4为本发明实施例1制得的最终产物的热重曲线图。
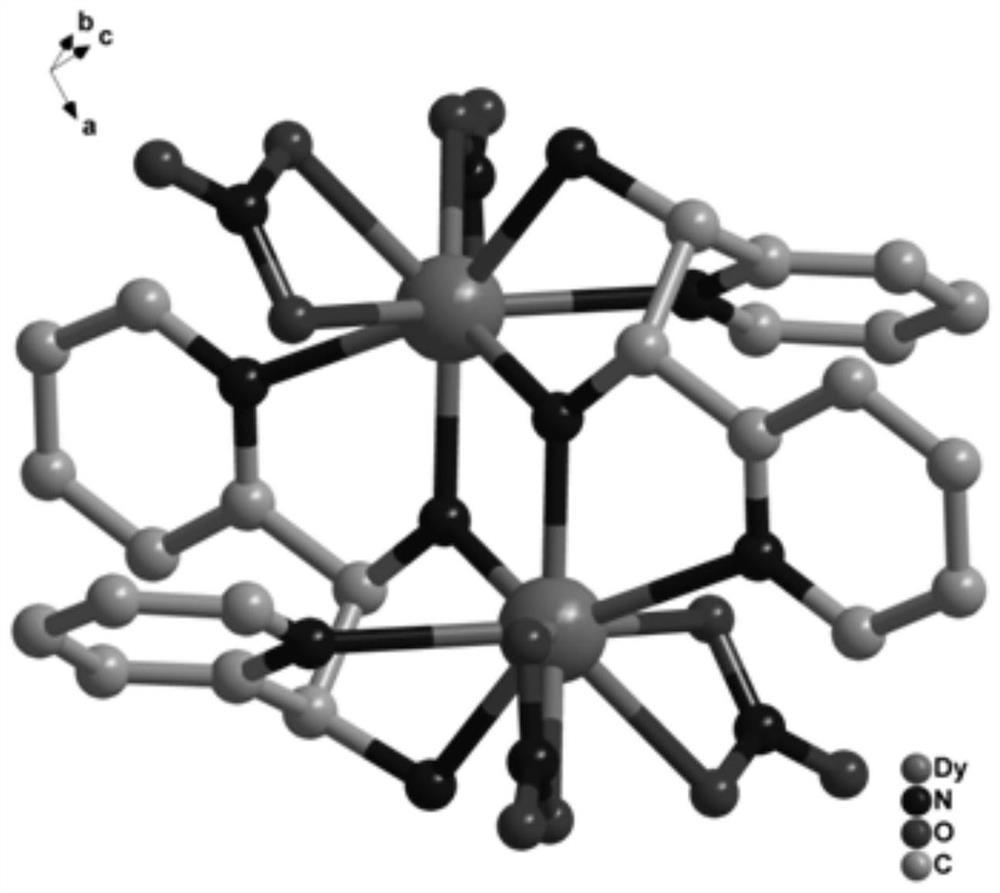
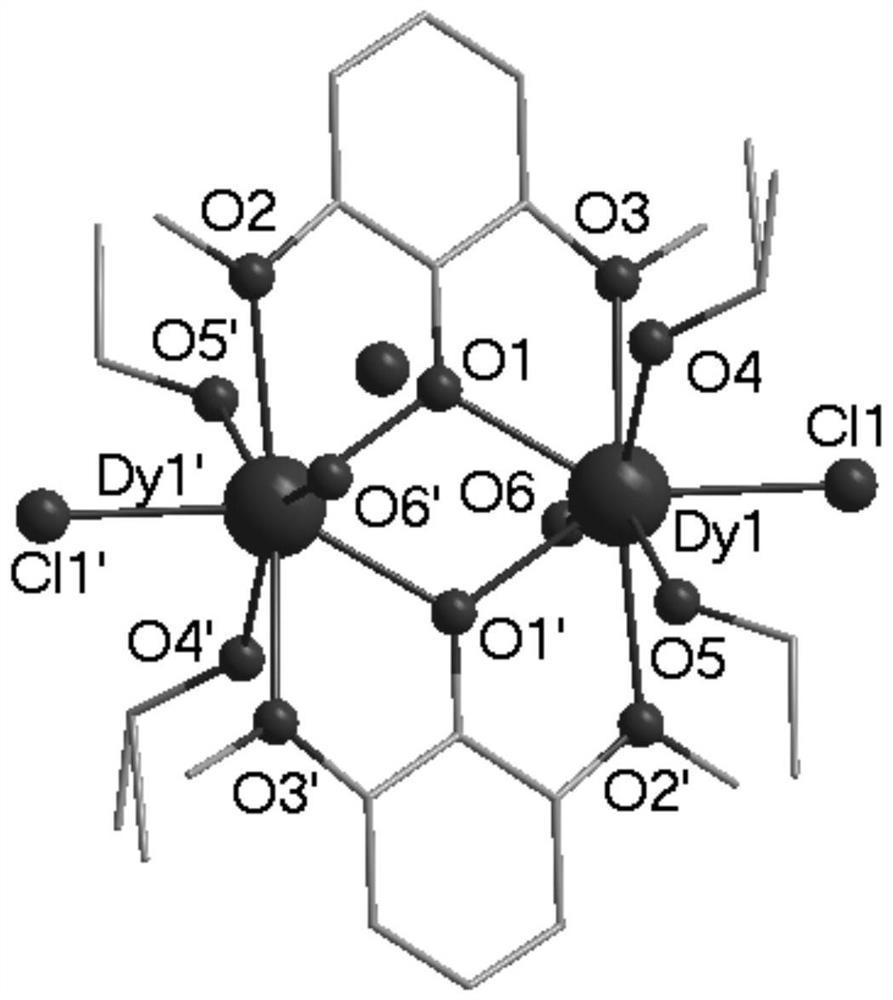
图5为本发明实施例1制得的最终产物的晶体结构图。
图6为对照的三组实验在与实施例1同样的条件下反应48h后,冷却后的内衬管及其中的反应物的照片;其中(a)表示HL2+Dy(NO3)3·6H2O,(b)表示HL1+Dy(NO3)3·6H2O,(c)表示HL1+HL2)。
图7为本发明所述配合物在不同溶剂中的紫外光谱图。
图8为本发明所述配合物在不同溶剂中的荧光光谱图。
图9为本发明所述配合物、HL1、HL2以及Dy(NO3)3·6H2O在乙醇中的荧光光谱图,图中complex表示本发明所述配合物。
图10为本发明所述配合物、HL1、HL2以及Dy(NO3)3·6H2O在乙腈中的荧光光谱图,图中complex表示本发明所述配合物。
具体实施方式
下面结合具体实施例对本发明作进一步的详述,以更好地理解本发明的内容,但本发明并不限于以下实施例。
实施例1:配合物[Dy2(L)2(NO3)4](以下也简称为配合物)的制备
称取0.5mmol 4-甲氧基-3-羟基苯甲醛(以下也简称为HL1)(76mg)置于带聚四氟乙烯内衬管的反应釜中,加入10mL由甲醇和乙腈按1:1的体积比组成的混合溶剂中,搅拌5min后溶解;再称取0.5mmol Dy(NO3)3·6H2O(228mg)置于反应釜中,继续搅拌5min后溶解;再取0.5mmol的2-(氨甲基)-吡啶(以下也简称为HL2或配体HL2)(61μL),充分搅拌5min后,使整个反应体系混合均匀后密闭;随后将反应釜置于温度为80℃下反应48h,取出,冷却,可观察到内衬管中有墨绿色长条状晶体析出(如图1所示),收集晶体,干燥。产率35%(基于HL2)。
对本实施例所得产物进行表征:
1)红外光谱分析,其谱图如图2所示。
由红外光谱图可看出:配合物在3400-3200cm-1之间出现了两个峰,分别是3273cm-1、3340cm-1我们知道伯胺和仲胺的伸缩震动吸收峰,伯胺呈两个在3200-3400cm-1之间,仲胺是一个,叔胺没有,很明显这两个峰对应于伯胺上的N-H伸缩振动;我们也知道3070-3020cm-1,1600-1510cm-1,900-700cm-1是吡啶的特征吸收峰,从图中可以看出1595cm-1、1042cm-1、786cm-1是吡啶的特征吸收峰;1600cm-1附近的吸收峰说明结构有-C=N。
2)粉末衍射分析,其谱图如图3所示。
由图3可知,实验谱图和根据晶体数据拟合得到的理论谱图基本吻合,证实配合物是纯相的,保证了表征得到的数据的可靠性。
3)热重分析,其曲线如图4所示。
热重分析显示,本发明所述配合物[Dy2(L)2(NO3)4]的热分析曲线在N2保护下,温度区间从30-1000℃,5℃/min的升温速度进行采集。配合物[Dy2(L)2(NO3)4]在260℃前质量相对较稳定,继续加热迅速失重。更高温度下框架发生热分解并伴随失重。从TG图可以看出,配合物[Dy2(L)2(NO3)4]中不存在客体分子。没有客体分子说明其分子间紧密堆积,同时也与其堆垛图中显示的堆垛方式相对应。
4)晶体结构分析:
通过X-射线衍射测定表面结构完好的墨绿色长条状晶体以确定其晶体结构,所得晶体结构数据如下述表1所示,部分键长数据如下述表2所示,部分键角数据如下述表3所示,所得墨绿色长条状晶体的晶体结构如图5所示,确定所得墨绿色长条状晶体为本发明所述的基于吡啶配体的双核镝配合物[Dy2(L)2(NO3)4],其中,L为2-氨基-1,2-二-吡啶-乙醇脱去羟基氢原子,带一个单位负电荷。
表1:本发明所述配合物的晶体结构数据表
表2:本发明所述配合物的部分键长表
表3:本发明所述配合物的部分键角表
X-射线单晶衍射显示,本发明所述配合物属于单斜晶系I2/a空间群,晶胞参数为 α=90.00°,β=110.003(3)°,γ=90.00°,不对称单元由一个配体HL2、两个NO3-和一个Dy(Ⅲ)组成。金属中心为九配位,由来自于配体的5个N原子和来自于硝酸根的4个O原子构成,两个镝离子间通过原位生成的配体上的氮原子桥连形成双核结构。
因单晶解析出来的结构中,HL1并未参与配位,本申请人随即做了三组对照实验(具体为(a)HL2+Dy(NO3)3·6H2O,(b)HL1+Dy(NO3)3·6H2O,(c)HL1+HL2),分别让它们在一样的实验条件下反应48h,结果显示,对照的三组实验都是澄清的溶液,并未出现沉淀或其他现象,冷却后的内衬管及其中的反应物的照片如图6所示。因此,本申请人推测HL1尽管没有参与配位,但可能在反应体系中,起到催化的作用。
实施例2
重复实施例1,不同的是,将甲醇和乙腈的体积比改为1:2。
结果得到墨绿色长条状晶体。产率28%(基于HL2)。
对本实施例所得产物经单晶衍射分析,确定所得墨绿色长条状晶体为本发明目标配合物。
实施例3
重复实施例1,不同的是,将甲醇和乙腈的体积比改为2:1,将反应改在120℃条件下进行,反应的时间为30h。
结果得到墨绿色长条状晶体。产率14%(基于HL2)。
对本实施例所得产物经单晶衍射分析,确定所得墨绿色长条状晶体为本发明目标配合物。
实施例4
重复实施例1,不同的是,将反应改在50℃条件下进行,反应的时间为60h。
结果得到墨绿色长条状晶体。产率19%(基于HL2)。
对本实施例所得产物经单晶衍射分析,确定所得墨绿色长条状晶体为本发明目标配合物。
实施例5:本发明所述配合物的紫外、荧光测定
将配合物分别溶解在不同的溶剂中,配置成浓度为1×10-5mol/mL的甲醇溶液(10mL)进行紫外和荧光测试。其紫外吸收峰如图7所示,结果显示所用溶剂的最大吸收峰在290nm左右有轻微的红移或者蓝移,本申请人以此吸收峰来测它的发射发,荧光测试条件为狭缝宽度设为5nm,扫速240nm/min,室温26度,扫描电压为490eV谱图如图8所示。
由图8可知:配合物在所用溶剂中的发射峰都在386nm、413nm、441nm、560nm左右且都有轻微的红移和蓝移,同时配合物在乙醇和乙腈的溶液中,荧光效果很好,荧光强度很高。为了检测是否为配体发光,本申请人分别配置了同等体积和浓度的本发明所述配合物、HL1、HL2和Dy(NO3)3·6H2O的乙醇及乙腈溶液,进行荧光测试,结果如图9和图10所示,测试条件为狭缝宽度设为5nm,扫速240nm/min,室温26度,扫描电压为490eV。
结果显示:HL1、HL2以及Dy(NO3)3·6H2O均未有荧光,由此可见:我们推测该化合物的发光属于金属-配体化合物的方式,而发光机理则被归结为配体到金属电荷转移引起的荧光发射(LMCT:ligand-to-metal charge transfer),本申请人由图推测,此配合物在工业上用于检测乙醇和乙腈具有潜在的价值。
基于吡啶配体的双核镝配合物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












![一种基于邻羧基苯乙酸配体的[CdNa]异金属荧光材料及其制备方法](https://www.zhichawang.com/images/ui/CN2019105683801/CN2019105683801.jpg)






动态评分
0.0