

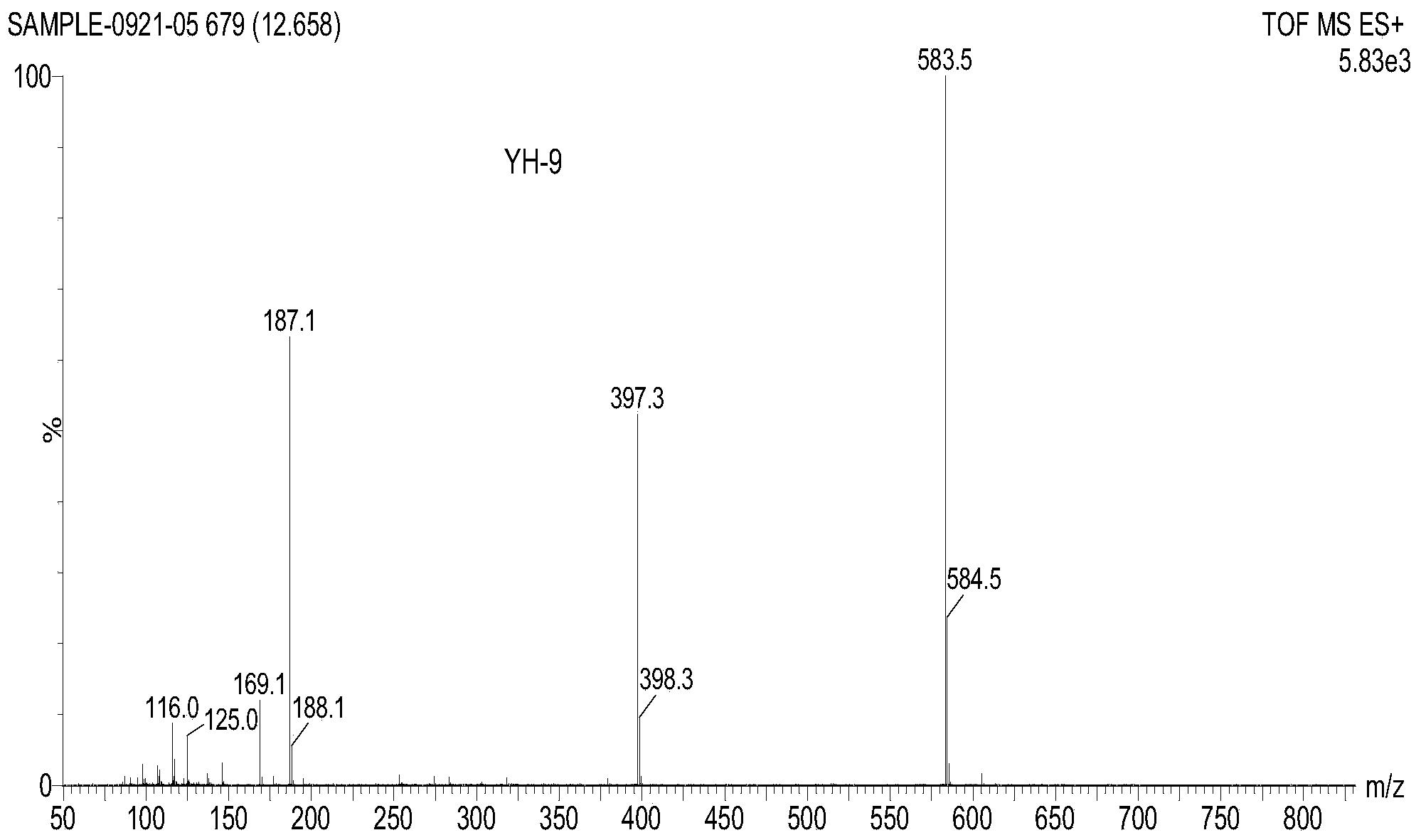





专利摘要
本发明提供了一种多取代氢化茚衍生物及其制备方法,与现有技术相比,本发明提供了一系列新的氢化茚衍生物。相对于普通氢化茚衍生物,本发明制备的氢化茚衍生物有多环的存在,其结构更加复杂多样,在化工生产中也将表现出更加广阔的用途前景。并且,本发明提供的制备方法简便、高效,反应时间短,效率高。
权利要求
1.一种多取代氢化茚衍生物的制备方法,其特征在于,所述制备方法包括以下步骤:
1)以氢化钠为催化剂,将丙二酸酯与炔丙基溴加入到无水乙腈中冰水浴,反应,然后纯化分离,得到黄棕色固体产物,即化合物1,结构式
2)将化合物1与苯乙炔基溴混合在Pd(PPh3)2Cl2/CuI的无水无氧催化体系中,以三乙胺作碱,以无水乙腈为溶剂,室温下搅拌反应,纯化分离后,得到浅黄色固体产物,即前体化合物2,结构式
3)步骤2)所制备的前体化合物2在甲苯溶剂中与2-丙二烯基-4,4,5,5-四甲基-1,3-二氧杂环戊硼烷反应,自然冷却至室温停止反应;将产物纯化分离,得到白色固体,即氢化茚衍生物;其结构式为:
其中E=CO2R,R为直链烷基、支链烷基或不饱和烃基。
2.一种多取代氢化茚衍生物的制备方法,其特征在于,所述制备方法包括以下步骤:
1)以氢化钠为催化剂,将丙二酸酯与炔丙基溴加入到无水乙腈中冰水浴,反应,然后纯化分离,得到黄棕色固体产物,即化合物1,结构式
2)将化合物1与对甲基苯乙炔基溴混合在Pd(PPh3)2Cl2/CuI的无水无氧催化体系中,以三乙胺作碱,以无水乙腈为溶剂,室温下搅拌反应,纯化分离后,得到浅黄色固体产物,即前体化合物2,结构式
3)步骤2)所制备的前体化合物2在甲苯溶剂中与2-丙二烯基-4,4,5,5-四甲基-1,3-二氧杂环戊硼烷反应,自然冷却至室温停止反应;将产物纯化分离,得到白色固体,即氢化茚衍生物;其结构式为:
其中E=CO2R,R为直链烷基、支链烷基或不饱和烃基。
3.根据权利要求1或2所述的制备方法,其特征在于,步骤1)中氢化钠、丙二酸酯、炔丙基溴与无水乙腈的摩尔比为4-5:1:2.2-3.2:20-23;所述丙二酸酯选自丙二酸二异丙酯。
4.根据权利要求1或2所述的制备方法,其特征在于,步骤1)中冰水浴条件下反应温度在0-5℃;反应时间在5小时以上。
5.根据权利要求1所述的制备方法,其特征在于,步骤2)中所述化合物1与苯乙炔基溴、Pd(PPh3)2Cl2/CuI、三乙胺和无水乙腈的物质的量比为1:2.2-3.2:0.03-0.04:4-5:30-45。
6.根据权利要求2所述的制备方法,其特征在于,步骤2)中所述化合物1与对甲基苯乙炔基溴、Pd(PPh3)2Cl2/CuI、三乙胺和无水乙腈的物质的量比为1:2.2-3.2:0.03-0.04:4-5:30-45。
7.根据权利要求1或2所述的制备方法,其特征在于,步骤2)所述搅拌反应,反应时间在10小时以上。
8.根据权利要求1或2所述的制备方法,其特征在于,步骤3)中前体化合物2、2-丙二烯基-4,4,5,5-四甲基-1,3-二氧杂环戊硼烷与甲苯的摩尔比的摩尔比为1:1:28-66。
9.根据权利要求1或2所述的制备方法,其特征在于,步骤3)中反应在95-100℃的条件下进行,时间12h以上。
10.根据权利要求1所述的制备方法,其特征在于,所述多取代氢化茚衍生物结构式为:
11.根据权利要求2所述的制备方法,其特征在于,所述多取代氢化茚衍生物结构式为:
说明书
技术领域
本发明属于有机化合物领域,具体涉及一种多取代氢化茚衍生物及其制备方法。
背景技术
茚在有机合成和生活中除用于有机溶剂和杀虫剂的中间体外,主要用于生产茚-古马隆树脂。其加氢还原产物茚满也用处广泛,可用作航空燃料的防震剂、橡胶工业防震剂,它的衍生物可制二十多种医药,例如抗艾滋病药CCR5拮抗剂、抗炎剂1-N-取代胺基-2肟基茚满盐酸盐闯等,还可用于有机合成工业,作溶剂使用,二氢茚经裂化可制苯类产品。
随着氢化茚衍生物的需求量与日俱增,高效便捷的合成方法逐渐显示出优势。
发明内容
本发明的目的在于提供一种多取代氢化茚衍生物的制备方法,简便、高效,反应时间短,效率高。
本发明还提供了一种多取代氢化茚衍生物,具有多环存在,结构更复杂,有广阔的应用前景。
本发明提供的一种多取代氢化茚衍生物的制备方法,包括以下步骤:
1)以氢化钠为催化剂,将丙二酸酯与炔丙基溴加入到无水乙腈中冰水浴,反应,然后纯化分离,得到黄棕色固体产物,即化合物1,结构式 R为直链烷基、支链烷基、饱和烃类、不饱和烃类或芳香烃类基团;
2)将化合物1与苯乙炔基溴或取代的苯乙炔基溴混合在Pd(PPh3)2Cl2/CuI的无水无氧催化体系中,以三乙胺作碱,以无水乙腈为溶剂,室温下搅拌反应,纯化分离后,得到浅黄色固体产物,即前体化合物2,结构式 R为直链烷基、支链烷基、饱和烃类、不饱和烃类或芳香烃类基团;
3)步骤2)所制备的前体化合物2在甲苯溶剂中与2-丙二烯基-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷反应,自然冷却至室温停止反应;将产物纯化分离,得到白色固体,即氢化茚衍生物。
进一步的,步骤1)中氢化钠、丙二酸酯、炔丙基溴与无水乙腈的摩尔比为4-5:1:2.2-3.2:20-23;所述丙二酸酯选自丙二酸二异丙酯。
步骤1)中冰水浴条件下反应温度在0-5℃;反应时间在5小时以上;
步骤1)中所述纯化分离具体为:产物加水洗涤,用乙酸乙酯萃取,减压旋干,得到黄棕色固体产物,即化合物1。
步骤2)中所述化合物1与苯乙炔基溴或取代的苯乙炔基溴、Pd(PPh3)2Cl2/CuI、三乙胺和无水乙腈的物质的量比为1:2.2-3.2:0.03-0.04:4-5:30-45;
步骤2)所述搅拌反应,反应时间在10小时以上;所述取代的苯乙炔基溴选自甲基苯乙炔基溴;
步骤2)中所述纯化分离具体为:产物用水洗涤,用乙酸乙酯萃取,减压旋干,用体积比为1:60的乙酸乙酯:石油醚柱层析分离,得到浅黄色固体产物,即前体化合物2。
步骤2)中所述Pd(PPh3)2Cl2/CuI的无水无氧催化体系中,摩尔比Pd(PPh3)2Cl2:CuI=3:1。
步骤3)中前体化合物2、2-丙二烯基-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷与甲苯的摩尔比的摩尔比为1:1:28-66;
步骤3)中反应在95-100℃的条件下进行,时间12h以上。
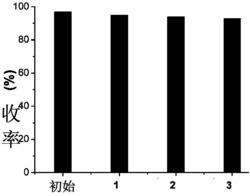
步骤3)中所述纯化分离具体为:将所得产物用水洗涤,乙酸乙酯萃取,减压旋干,用体积比1:40的乙酸乙酯:石油醚的柱层析分离,得到白色固体,即氢化茚衍生物。柱层析产率约为76.1%。
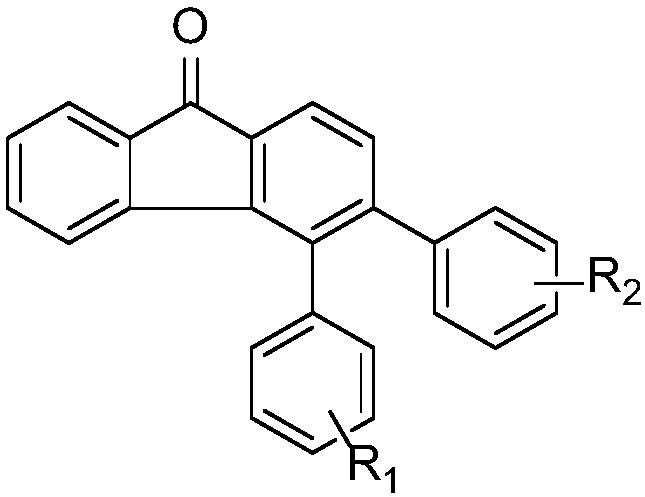
本发明提供的一种多取代氢化茚衍生物采用上述方法制备得到,结构式为:
其中E=CO2R,R为直链烷基、支链烷基、饱和烃类、不饱和烃类或芳香烃类基团;R1为卤素、直链烷基、支链烷基、酯基、烷氧基以及其相应的衍生物。
优选的,多取代氢化茚衍生物结构式为: 或
与现有的氢化茚衍生物(例如1-N-取代胺基-2肟基茚满盐酸盐闯、1-甲氧基-2-羟基-茚满)相比,本发明提供了一系列新的氢化茚衍生物。相对于普通氢化茚衍生物,本发明制备的氢化茚衍生物有多环的存在,其结构更加复杂多样。氢化茚衍生物在医药工业中应用广泛,例如抗艾滋病药CCR5拮抗剂、抗炎剂1-N-取代胺基-2肟基茚满盐酸盐闯等,本发明物质可能在在医药工业中也能具有广泛作用,并且本发明提供的制备方法简便、高效,反应时间短,效率高。
附图说明
图1为氢化茚衍生物的结构式;
图2为实施例1制备的氢化茚衍生物的结构式;
图3为实施例2制备的氢化茚衍生物的结构式;
图4为实施例1制备的氢化茚衍生物的核磁共振氢谱;
图5为实施例1制备的氢化茚衍生物的核磁共振碳谱;
图6为实施例2制备的氢化茚衍生物的核磁共振氢谱;
图7为实施例2制备的氢化茚衍生物的核磁共振碳谱;
图8为实施例1制备过程反应方程式;
图9为实施例2制备过程反应方程式;
图10为实施例1步骤3的反应原理示意图。
具体实施方式
实施例1
一种氢化茚衍生物,所述的氢化茚衍生物结构式为:
一种多取代氢化茚衍生物的制备方法,所述的制备方法包括以下步骤:
(1)以830mmol氢化钠为催化剂,将200mmol丙二酸二异丙酯与440mmol炔丙基溴加入到210mL无水乙腈中冰水浴,搅拌反应8小时,产物加水洗涤,用乙酸乙酯萃取,减压旋干,得到棕黄色固体产物,即化合物1;
(2)将80mmol化合物1与200mmol苯乙炔基溴混合在Pd(PPh3)2Cl2/CuI的无水无氧催化体系中(2.56mmol/0.85mmol),摩尔比Pd(PPh3)2Cl2:CuI=3:1,以336mmol三乙胺作碱,以150mL无水乙腈为溶剂,室温下搅拌反应12小时,产物用水洗涤,用乙酸乙酯萃取,减压旋干,用体积比为1:60的乙酸乙酯:石油醚柱层析分离,得到浅黄色固体产物,即前体化合物2。
(3)在100℃的条件下,步骤(2)所制备的1mmol前体化合物2在5mL甲苯溶剂与1mmol 2-丙二烯基-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷反应18小时,得化合物3,即氢化茚衍生物的粗产物;将制备的氢化茚衍生物的粗产物用水洗涤,乙酸乙酯萃取,减压旋干,用体积比乙酸乙酯:石油醚=1:40柱层析分离,得到白色固体产物,即氢化茚衍生物,柱层析产率约为76.1%。
白色固体产物结构通过;
白色固体产物:
实施例2
一种氢化茚衍生物,所述的氢化茚衍生物结构式为:
一种多取代氢化茚衍生物的制备方法,所述的制备方法包括以下步骤:
(1)以氢830mmol化钠为催化剂,将200mmol丙二酸二异丙酯与440mmol炔丙基溴加入到210mL无水乙腈中冰水浴,搅拌反应8小时,产物加水洗涤,用乙酸乙酯萃取,减压旋干,得到棕黄色固体产物,即化合物1;
(2)将80mmol化合物1与200mmol对甲基苯乙炔基溴混合在Pd(PPh3)2Cl2/CuI的无水无氧催化体系中(2.56mmol/0.85mmol),摩尔比Pd(PPh3)2Cl2:CuI=3:1,以336mmol三乙胺作碱,150ml无水乙腈为溶剂,室温下搅拌反应12小时,产物用水洗涤,用乙酸乙酯萃取,减压旋干,用体积比为1:60的乙酸乙酯:石油醚柱层析分离,得到浅黄色固体产物,即前体化合物2(图9)。
(3)在100℃的条件下,步骤(2)所制备的1mmol前体化合物4在5mL甲苯溶剂与1mmol2-丙二烯基-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷反应18小时,得化合物3(图9),即氢化茚衍生物的粗产物;将制备的氢化茚衍生物的粗产物用水洗涤,乙酸乙酯萃取,减压旋干,用体积比乙酸乙酯:石油醚=1:40柱层析分离,得到白色固体产物,即氢化茚衍生物,柱层析产率约为75.7%。
白色固体产物结构通过;
白色固体产物:
一种多取代氢化茚衍生物及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。








![一种制备螺[环丙烷-1,2’-茚]-1’,3’-二酮化合物的方法](https://www.zhichawang.com/images/CN110028407A/CN110028407A.jpg)


动态评分
0.0