








专利摘要
本发明公开了一种以2‑氨基‑5‑氯二苯甲酮缩氨基硫脲为配体的铂配合物及其合成方法和应用,所述铂配合物的合成方法为:1)将2‑氨基‑5‑氯二苯甲酮溶于乙醇,再加入氨基硫脲混合均匀后,滴加3‑4滴浓硫酸,回流、过滤,滤液室温挥发,有淡黄色晶体析出,晶体过滤,无水乙醇洗涤2‑3次得化合物;2)取步骤1)得到的化合物和Pt(DMSO)2Cl2置于一端密封的玻璃管中,滴加甲醇至玻璃管中溶解后,真空密封玻璃管,将玻璃管置于60℃鼓风干燥箱中静置72h,得到目标配合物。该配合物结构新颖,活性大大提高,具有更加优良的抑神经瘤活性,又利于穿透BBB,以及一定程度上克服了药物耐药性。
权利要求
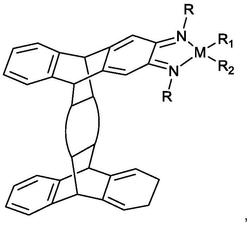
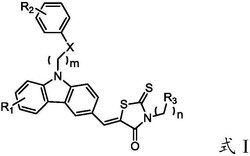
1.一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物,其特征在于,所述铂配合物的结构式如下所示:
所述铂配合物的合成路线为:
所述C1-C2、C4所示的铂配合物的合成方法,包括如下步骤:
1)将2-氨基-5-氯二苯甲酮溶于乙醇,再加入氨基硫脲混合均匀后,滴加3-4滴浓硫酸,回流、过滤,滤液室温挥发,有淡黄色晶体析出,晶体过滤,无水乙醇洗涤2-3次得化合物;
2)取步骤1)得到的化合物和Pt(DMSO)2Cl2置于一端密封的玻璃管中,滴加甲醇或DMF和CH3OH至玻璃管中溶解后,真空密封玻璃管,将玻璃管置于60℃鼓风干燥箱中静置72h,得到目标配合物;
步骤1)中,所述的氨基硫脲与2-氨基-5-氯二苯甲酮的物质的量之比为1:1;氨基硫脲与乙醇的物质的量之比为3:3500。
2.根据权利要求1所述的一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物,其特征在于,所述的氨基硫脲,为4-甲基-3-氨基硫脲、(2-甲基苯基)-3-氨基硫脲、氨基硫脲和4-(N,N-四氢吡咯)-3-氨基硫脲中的一种。
3.根据权利要求1所述的一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物,其特征在于,步骤1)中,所述的回流,回流温度为65℃,回流时间为4h。
4.根据权利要求1所述的一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物,其特征在于,步骤2)中,所述的化合物与Pt(DMSO)2Cl2的物质的量之比为1:1;化合物与甲醇或DMF和CH3OH的物质的量之比为1:0.9-1。
5.如权利要求1所述的铂配合物为活性成分在制备抗肿瘤药物中的应用。
说明书
技术领域
本发明涉及金属配合物的合成,具体是一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物及其合成方法和应用。
背景技术
血脑屏障(Blood Brain Barrier,BBB)是中枢神经系统与循环系统分离的动态界面。它通过调节中枢神经系统和循环系统之间的物质交换来维持体内平衡和正常的神经功能。据研究报导,只有不到3%的中枢神经系统药物可穿过BBB达到治疗目的,而且基本为小分子药物。因此,对于药物研发企业而言开发新的脑部疾病药物的风险非常大,这极大地限制了脑部药物的开发。特别是脑肿瘤的治疗仍然被BBB所限制。化合物必须通过BBB进入脑组织,这已成为开发治疗脑肿瘤化合物的首要问题。本发明通过参考上市多年的神经类药物小分子,由于其良好的穿透BBB能力,为抑制脑肿瘤化合物的设计提供了新的思路。为实现体内外对脑肿瘤良好抑制活性,以及脂溶性神经药物小基团增强化合物穿透BBB能力的双重作用,我们利用了苯二氮卓类镇静催眠药作为神经药物小分子的特殊结构片段(2-氨基-5-氯二苯甲酮)。而通过BBB的关键片段2-氨基-5-氯二苯甲酮,它主要作为无毒产物被肝脏代谢。然而,2-氨基-5-氯二苯甲酮没有明显的抗肿瘤活性。幸运的是,由于其特殊的结构能与氨基硫脲缩合成席夫碱,进而可以合成相关金属配合物,不仅能够增强抗肿瘤活性还能够提高化合物的靶向性。
氨基硫脲作为抗肿瘤药物已有半个多世纪的历史,主要机制包括直接诱导DNA损伤和抑制拓扑异构酶Ⅱ。在过去的半个世纪中,对硫代氨基脲进行了很多抗肿瘤的研究。自从Rosenberg发现顺铂(cis-[PtCl2(NH3)2])具有抗肿瘤活性以来,金属-有机配体配合物的研究掀起了研究热潮。铂类药物主要通过与DNA结合,引起交叉联结,从而破坏DNA的功能,抑制肿瘤细胞生长。研究表明,金属-有机配体复合物比单独配体具有更高的生物活性,特别是在抗肿瘤活性和降低金属的毒性方面。
因此,为实现化合物不仅能够抑制神经瘤细胞,而且又利于穿透BBB。本本发明合成了系列全新的2-氨基-5-氯二苯甲酮缩氨基硫脲铂金属配合物以及对其抑神经瘤活性进行了研究。
发明内容
本发明的目的在于提供一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物及其合成方法和应用,该配合物结构新颖,活性大大提高,具有更加优良的抑神经瘤活性,又利于穿透BBB,以及一定程度上克服了药物耐药性。
实现本发明目的的技术方案是:
一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物,所述铂配合物的结构式如下C1-C4所示:
上述式C1-C4所示铂配合物的合成路线为:
上述式C1-C4所示铂配合物的合成方法,包括如下步骤:
1)将2-氨基-5-氯二苯甲酮溶于乙醇,再加入氨基硫脲混合均匀后,滴加3-4滴浓硫酸,回流、过滤,滤液室温挥发,有淡黄色晶体析出,晶体过滤,无水乙醇洗涤2-3次得化合物;
2)取步骤1)得到的化合物和Pt(DMSO)2Cl2置于一端密封的玻璃管中,滴加甲醇至玻璃管中溶解后,真空密封玻璃管,将玻璃管置于60℃鼓风干燥箱中静置72h,得到目标配合物。
步骤1)中,所述的氨基硫脲与2-氨基-5-氯二苯甲酮的物质的量之比为1:1;氨基硫脲与乙醇的物质之比为3:3500。
所述的氨基硫脲,为4-甲基-3-氨基硫脲、(2-甲基苯基)-3-氨基硫脲、氨基硫脲和4-(N,N-四氢吡咯)-3-氨基硫脲中的一种。
步骤1)中,所述的回流,回流温度为65℃,回流时间为4h。
步骤2)中,所述的化合物与Pt(DMSO)2Cl2的物质的量之比为1:1;化合物与甲醇的物质的量之比为1:0.9-1。
本发明进一步包括以铂配合物C1-C4为活性成分制备的抗肿瘤药物。
有益效果:本发明提供的一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物及其合成方法和应用,与现有技术相比,该配合物结构新颖,活性大大提高,具有更加优良的抑神经瘤活性,又利于穿透BBB,以及一定程度上克服了药物耐药性。
附图说明
图1为实施例1合成的C1配合物的单晶结构图;
图2为实施例2合成的C2配合物的单晶结构图;
图3为实施例3合成的C3配合物的单晶结构图;
图4为实施例4合成的C4配合物的单晶结构图。
具体实施方式
下面结合附图和实施例对本发明做进一步阐述,但不是对本发明的限定。
实施例1:
C1配合物的合成方法为:
1)将3mmol(693mg)2-氨基-5-氯二苯甲酮溶于20mL乙醇,再加入3mmol(315mg)4-甲基-3-氨基硫脲混合均匀后,滴加3-4滴浓硫酸,回流、过滤,滤液室温挥发,有淡黄色晶体析出,晶体过滤,无水乙醇洗涤2-3次得化合物1;
Yield:0.75g,78.6%,light yellow solid;Rf=0.414(Petroleum ether:EtOAc=1:1).M.p.133-135℃.1H NMR(400MHz,DMSO-d6)δ8.79(q,J=4.5Hz,1H),8.70(s,1H),7.72-7.69(m,2H),7.42-7.37(m,3H),7.29(dd,J=8.8,2.5Hz,1H),6.92-6.90(m,2H),5.28(s,2H),3.06(d,J=4.6Hz,3H).13C NMR(100MHz,DMSO-d6)δ177.72,145.97,144.55,135.86,130.55,129.54,128.25,127.55,119.68,117.44,116.18,31.30.ESI+m/z:calcd for C15H16ClN4S,317[M-H]+。
2)取步骤1)得到的0.05mmol(15.9mg)化合物1和23mg Pt(DMSO)2Cl2置于一端密封的玻璃管中,滴加2mL CH3OH至玻璃管中溶解后,真空密封玻璃管,将玻璃管置于60℃鼓风干燥箱中静置72h,得到目标配合物C1,其单晶结构如图1所示。
Yield:0.04g,71.5%,IR,cm-1:IR,cm-1:3434(s,amide),2926(m,aromatic hydrogen),1616(s),1524(s,-C=C-),1384(s),1262(s,thioamide),1186(s),823(s),743(s,ph-H).ESI+m/z:calcd for C18H20AuClN3OS,558[M+H]+。
实施例2:
C2配合物的合成方法为:
1)将3mmol(693mg)2-氨基-5-氯二苯甲酮溶于20mL乙醇,再加入3mmol(543mg)4-(2-甲基苯基)-3-氨基硫脲混合均匀后,滴加3-4滴浓硫酸,回流、过滤,滤液室温挥发,有淡黄色晶体析出,晶体过滤,无水乙醇洗涤2-3次得化合物2;
Yield:0.71g,60.0%,light yellow solid;Rf=0.421(Petroleum ether:EtOAc=2:
1).M.p.140-142℃.1H NMR(400MHz,DMSO-d6)δ10.23(s,1H),9.04(s,1H),7.82-7.79(m,2H),7.43-7.37(m,3H),7.33-7.29(m,3H),7.25-7.22(m,2H),6.99-6.93(m,2H),5.39(s,2H),2.27(s,3H).13C NMR(100MHz,DMSO-d6)δ176.99,146.83,144.63,137.91,135.82,135.24,130.63,130.08,129.68,128.40,128.29,128.23,127.88,126.83,125.93,119.76,117.56,116.40,17.75.ESI+m/z:calcd for C21H18ClN4S,393[M-H]+。
2)取步骤1)得到的0.05mmol(15.2mg)化合物2和23mg Pt(DMSO)2Cl2置于一端密封的玻璃管中,滴加2mL体积比为1:1的DMF和CH3OH混合溶液至玻璃管中溶解后,真空密封玻璃管,将玻璃管置于60℃鼓风干燥箱中静置72h,得到目标配合物C2,其单晶结构如图2所示。
IR,cm-1:3422(s,amide),1619(s),1571(s,-C=C-),1521(s,-C=C-),1488(s,-C=C-),1248(m,thioamide),1155(s),866(s),816(s,ph-H),771(s,ph-H),736(s,ph-H).ESI+m/z:calcd for C42H35Cl6N8Pt2S2,1314[M+H]+。
实施例3:
C3配合物的合成方法为:
1)将3mmol(693mg)2-氨基-5-氯二苯甲酮溶于20mL乙醇,再加入3mmol(273mg)氨基硫脲混合均匀后,滴加3-4滴浓硫酸,回流、过滤,滤液室温挥发,有淡黄色晶体析出,晶体过滤,无水乙醇洗涤2-3次得化合物3;
Yield:0.76g,83.3%,light yellow solid;Rf=0.321(Petroleum ether:EtOAc=1:1).M.p.158-160℃.1H NMR(400MHz,DMSO-d6)δ8.61(s,2H),8.28(s,1H),7.72-7.69(m,2H),7.41-7.35(m,3H),7.28(dd,J=8.8,2.6Hz,1H),6.92-6.90(m,2H),5.29(s,2H).13C NMR(100MHz,DMSO-d6)δ178.09,146.40,144.57,135.80,130.57,129.60,128.27,127.63,119.68,117.45,116.12.ESI+m/z:calcd for C14H14ClN4S,303[M-H]+。
2)取步骤1)得到的0.05mmol(17.9mg)化合物3和23mg Pt(DMSO)2Cl2置于一端密封的玻璃管中,滴加2mL体积比为1:1的DMF和CH2Cl2混合溶液至玻璃管中溶解后,真空密封玻璃管,将玻璃管置于60℃鼓风干燥箱中静置72h,得到目标配合物C3,其单晶结构如图4所示。
IR,cm-1:3439(s,amide),1630(s),1549(s,-C=C-),1517(s,-C=C-),1452(s,-C=C-),1268(m,thioamide),1150(s),820(s),775(s,ph-H),756(s,ph-H),735(s,ph-H).ESI+m/z:calcd for C16H18ClN4OPtS2,576[1/4M+DMSO+H]+。
实施例4:
C4配合物的合成方法为:
1)将3mmol(693mg)2-氨基-5-氯二苯甲酮溶于20mL乙醇,再加入3mmol(435mg)4-(N,N-四氢吡咯)-3-氨基硫脲混合均匀后,滴加3-4滴浓硫酸,回流、过滤,滤液室温挥发,有淡黄色晶体析出,晶体过滤,无水乙醇洗涤2-3次得化合物4;
Yield:0.65g,60.5%,light yellow solid;Rf=0.35(Petroleum ether:EtOAc=1:1).M.p.135-137℃.1H NMR(400MHz,DMSO-d6)δ8.65(s,1H),7.71-7.69(m,2H),7.42-7.38(m,3H),7.307.27(m,1H),6.92-6.90(m,2H),5.29(s,2H),3.59(q,J=6.8Hz,2H),1.64-1.56(m,2H),1.38-1.29(s,2H),0.94-0.90(m,2H).13C NMR(100MHz,DMSO-d6)δ176.91,146.05,144.56,135.85,130.55,129.54,128.26,127.56,119.68,117.44,116.22,43.77,30.82.ESI+m/z:calcd for C18H18ClN4S,357[M-H]+。
2)取步骤1)得到的0.05mmol(19.7mg)化合物4和23mg Pt(DMSO)2Cl2置于一端密封的玻璃管中,滴加2mL体积比为1:1的DMF和CH3OH混合溶液至玻璃管中溶解后,真空密封玻璃管,将玻璃管置于60℃鼓风干燥箱中静置72h,得到目标配合物C4,其单晶结构如图4所示。
IR,cm-1:3452(s,amide),1628(s),1536(s,-C=C-),1517(s,-C=C-),1452(s,-C=C-),1260(m,thioamide),1161(s),813(s),769(s,ph-H),734(s,ph-H).ESI+m/z:calcd for C20H24ClN4OPtS2,630[1/4M+DMSO+H]+。
体外活性测试
对进行了体外增殖抑制活性实验:
1、细胞株与细胞培养
本实验选用了人神经瘤细胞株(SK-N-MC),人非小肺癌细胞(A549),人胃癌细胞(MGC-803),人正常肝细胞(HL-7702)进行了活性探究。
所有细胞株均培养在含10%小牛血清、100U/mL链霉素的RPMI-1640/DMEM培养液内,置37℃含体积浓度5%CO2孵箱中培养。
2、待测化合物的配制所用的受试药物的纯度≥95%,将其DMSO储液用生理缓冲液稀释后配置成5mmol/L的终溶液,其中助溶剂DMSO的浓度≤1%,测试该浓度下化合物对各种肿瘤细胞生长得抑制程度。
3、细胞生长抑制实验(MTT法)
(1)取对数生长期的肿瘤细胞,经胰蛋白酶消化后,用含10%小牛血清的培养液配置成浓度为5000个/mL的细胞悬液,以没孔180μL接种于96孔培养板中,使待测细胞浓度至每孔1000~10000/孔(边缘孔用无菌PBS填充);
(2)5%CO2,37℃孵育24h,至细胞单层铺满孔底,每孔加入一定浓度梯度的药物20μL,每个浓度梯度设5个复孔;
(3)5%CO2,37℃孵育48h,至倒置显微镜下观察;
(4)每孔加入10μL的MTT溶液(5mg/mL PBS,即0.5%MTT),继续培养4h-6h;
(5)终止培养,小心吸去孔内培养液,每孔加入100μL的DMSO充分溶解甲瓒沉淀,振荡器混匀后,在酶标仪用波长为570nm,参比波长为450nm测定各孔的光密度值;
(6)根据测得的光密度值(OD值),来判断活细胞数量,OD值越大,细胞活性越强。利用公式:
肿瘤细胞生长抑制率(%)=[(1-实验组平均OD值)/(对照组平均OD值)]×%;
IC50测定:利用以上方法,每种化合物须设置浓度梯度,其中含多个(一般5~8个)浓度,每个浓度也须设置3~5个副孔,实验得到每个不同浓度的抑制率,然后在SPSS软件中计算化合物的IC50值。
表1各化合物对不同细胞株的IC50值(μM)
在表1中,IC50值(μM)越低表明化合物抑制活性越好。本实验研究结果表明,对所测试的几种肿瘤细胞株,合成的系列2-氨基-5-氯二苯甲酮缩氨基硫脲铂配合物对人神经瘤细胞具有高度特异性,表现出很好的抑制活性,而且这些铂配合物对人正常肝细胞也没有明显毒性。
一种以2-氨基-5-氯二苯甲酮缩氨基硫脲为配体的铂配合物及其合成方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0