







专利摘要
本发明提出了一种吡咯衍生物的合成方法,其是在反应容器中按摩尔比1:1~1.4:1.3~4依次加入胺、吸电子炔烃、烯烃,按1mmol胺加入2~4mL的溶剂的比例加入溶剂,接着加入胺摩尔量0.8%~2%的催化剂Pd(OAc)2、胺摩尔量1.8%~4%的氧化剂K2S2O8、醋酸2mL、已腈2mL,在100℃~120℃油浴条件下反应15~24h,冷却至室温,加水,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,得到产品吡咯衍生物。本发明具有反应操作简单,反应底物廉价,产率高、选择性好、易分离纯化、污染少的特点。
权利要求
1.一种吡咯衍生物的合成方法,其特征是包括以下步骤:
在反应容器中按摩尔比1:1:1.3~4依次加入胺Ⅰ、吸电子炔烃Ⅱ、烯烃Ⅲ,接着加入胺摩尔量5%的催化剂Pd(OAc)2、胺摩尔量 2倍当量的氧化剂K2S2O8、醋酸 2 mL、溶剂乙腈2mL,在100℃~120℃油浴条件下反应15~24h,冷却至室温,加水,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,得到产品吡咯衍生物;该合成方法的反应通式为:
其中: R1为苯基、邻甲基苯基、邻碘苯基、间甲基苯基、间氟苯基、间氯苯基、间三氟甲基苯基、对甲基苯基、对甲氧基苯基、对氟苯基、对氯苯基、对溴苯基、对三氟甲基苯基、2,6-二甲基苯基、2,4-二甲基苯基、环己基或苄基;
所述胺I与溶剂的比例为1 mmol:2 mL。
2.根据权利要求1所述的一种吡咯衍生物的合成方法,其特征是所述的胺Ⅰ为苯胺,吸电子炔烃Ⅱ为丁炔二酸二甲酯,烯烃Ⅲ为苯乙烯,溶剂为乙腈。
3.根据权利要求1所述的一种吡咯衍生物的合成方法,其特征是所述的柱层析条件为:300~400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:20。
说明书
技术领域
本发明涉及吡咯衍生物,具体涉及一种吡咯衍生物的合成方法。
背景技术
吡咯和它的衍生物具有胺的一些性质,在以下几个方面具有很广的应用:1.吡咯及其衍生物是重要的医药中间体,可以参与合成许多药物。如酒石酸右吗拉胺、吡咯卡因、普环定等。2.吡咯及其衍生物也是合成农药、杀虫剂及杀菌剂的一种重要中间体。3.其衍生物还广泛应用于高分子中、新的材料、食品等领域。由于吡咯衍生物具有很广的应用前景和特别的一些生物活性,比如有重要的生理和药理活性,具有抗疟疾和抗菌等其他生物活性,从而人们对于其的研究也越来越深入。主要合成吡咯衍生物的方法有以下三种:1.Knorr吡咯合成方法(J.Am.Chem.Soc.1988,110,2992,Chem.Rev.2013,113,3084,Chem.Soc.Rev.2010,39,4402),此方法利用α-氨基酮或α-氨基酯和具有更强的α-活波氢的酮酯或二酮类化合物进行缩合反应,生成吡咯及其衍生物。为了避免异构体的产生,一般都选用对称的二酮类化合物作为起始原料,同时为了防止氨基酮的自身缩合,氨基酮可以替换为氨基肟或α-氨基酰胺作原料,从而提高反应的选择性。这个法的优点是它的合成路线较简单,但缺点是反应的产率低,选择性也不好;2.Paal-Knorr反应(Chem.Rev.2008,108,264,Chem.Commun.2016,52,6253),Pall在Knorr合成法的基础之上利用1,4-二羰基化合物与伯胺为起始原料脱水生成吡咯及其衍生物。此法虽简单易操作,但是产率一般;3.多组分一锅法(Chem.Soc.Rev.1991,20,391),此法是指在一个合成操作中,将至少两种以上的原料加入到一个反应器中,生成一类新化合物,这种化合物包括底物的主要结构片段,此法虽操作简单,但产率一般,适用范围小。近年来,吡咯衍生物在新型药物创造中有具有重要地位,其合成方法的研究也有很大的进展,2004年,宋智泉使用2-羰基肟与炔烃进行烷基化反应(化学通报,2006,04,306),同时发生分子内的Michael加成反应,从而生成吡咯衍生物,这种方法虽然简单但是产率不高。2008年,刘静等人采用苯胺和乙二醇直接气相合成吡咯(催化学报2008,02,159)。又如Chandrashaker等人用对称的β-二酮类化合物和α-氨基酮为原料合成了吡咯衍生物(Tetrahedron,2012,68,6957),此反应的缺点是产率低,选择性也低。2010年,Vivek等人在140℃的微波条件下加热(Angew.Chem.,Int.Ed.2016,55),用水做溶剂,纳米有机物做催化剂合成了吡咯衍生物的合成。但是此反应温度高,容易导致目标产物的分解。这些合成方法存在很多缺点:反应条件苛刻,反应温度高,压力高,分离困难,反应过程需要酸的量太大,易于腐蚀设备,对设备的要求高,反应的底物限制性较强,合成的吡咯衍生物的结构单一。
发明内容
针对现有吡咯衍生物的合成方法所存在的缺陷,本发明所要解决的技术问题是提供一种吡咯衍生物的合成方法,该合成方法操作简单,所用原料便宜,并且产率高、选择性好。
为解决上述技术问题,本发明所采取的技术方案是:一种吡咯衍生物的合成方法,包括以下步骤:
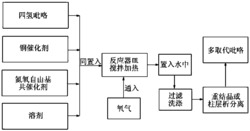
在反应容器中按摩尔比1:1~1.4:1.3~4依次加入胺Ⅰ、吸电子炔烃Ⅱ、烯烃Ⅲ,按1mmol胺加入2~4mL的溶剂的比例加入溶剂,接着加入胺摩尔量0.8%~2%的催化剂Pd(OAc)2、胺摩尔量1.8%~4%的氧化剂K2S2O8、醋酸2mL、已腈2mL,在100℃~120℃油浴条件下反应15~24h,冷却至室温,加水,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,得到产品吡咯衍生物;该合成方法的反应通式为:
其中,吡咯衍生物的化学结构通式如下:
上述结构式中,R1为C1~C6链状烷基、噻吩基、吡咯基、环己基或芳基;R2为-H、苯基或酯基;R3为酯基;R4为C3~C6链状烷基、噻吩基、吡咯基、环己基或芳基。
所述的胺Ⅰ为苯胺、邻氟苯胺、间氟苯胺、对氟苯胺、邻氯苯胺、间氯苯胺、对氯苯胺、邻溴苯胺、间溴苯胺、对溴苯胺、邻碘苯胺、邻硝基苯胺、间硝基苯胺、对硝基苯胺、邻甲氧基苯胺、间甲氧基苯胺、对甲氧基苯胺、邻甲基苯胺、间甲基苯胺、对甲基苯胺、邻三氟苯胺、间三氟苯胺、对三氟苯胺、2-苯基乙胺、3-苯基乙胺、4-苯基乙胺、2-噻吩甲胺、2-呋喃甲胺或者正己胺。
所述的吸电子炔烃Ⅱ为丁炔二酸二甲酯、丁炔二酸二乙酯、苯丙炔酸甲酯、苯丙炔酸乙酯、丙炔酸甲酯、丁炔酸甲酯、丙炔酸乙酯或者丁炔酸乙酯。
所述的烯烃Ⅲ为苯乙烯、邻氟苯乙烯、间氟苯乙烯、对氟苯乙烯、邻氯苯乙烯、间氯苯乙烯、对氯苯乙烯、邻溴苯乙烯、间溴苯乙烯、对溴苯乙烯、邻甲氧基苯乙烯、间甲氧基苯乙烯、对甲氧基苯乙烯、邻甲基苯乙烯、间甲基苯乙烯、对甲基苯乙烯、邻三氟苯乙烯、间三氟苯乙烯、对三氟苯乙烯、2-乙烯基噻吩或者2-乙烯基吡咯。
所述的溶剂为已腈、THF或者1,2-二氯乙烷。
所述的柱层析条件为:300~400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:20。
本方面采用上述技术方案所产生的有益效果是:
1.本发明利用烯烃代替传统原料炔烃的使用,反应操作比较简单;
2.反应底物简单,来源广泛,适合各种官能团取代的胺(包括邻位大位阻的官能团取代的苯胺),立体效应对反应的影响小;
3.本发明反应底物廉价,产率高、选择性好、易分离纯化、污染少,步骤简单,可以省略官能团的保护和去保护合成步骤,目标产物吡咯衍生物广泛适用于有机化学反应的配体、药物中间体和光电材料方面。
附图说明
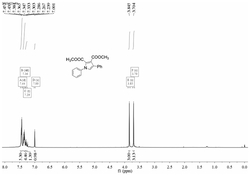
图1表示化合物1,4-二苯基-2,3-二甲酸甲酯基吡咯的1H NMR表征图;
图2表示化合物1,4-二苯基-2,3-二甲酸甲酯基吡咯的13C NMR表征图;
图3表示化合物1-对溴苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的1H NMR表征图;
图4表示化合物1-对溴苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的13C NMR表征图。
具体实施方式
下面结合实施例对本发明一种吡咯衍生物的合成方法作具体说明。
实施例1
一种1,4-二苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(93mg)的苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4a)298.2mg,产率89%,纯度99.9%。1H NMR(500MHz,CDCl3)ppm:8.43(d,J=8.0Hz,1H),8.34(d,J=8.0Hz,2H),8.00(d,J=8.5Hz,1H),7.92(s,1H),7.80(t,1H),7.51-7.64(m,9H);13C NMR(500MHz,CDCl3):156.90,149.23,149.05,139.77,138.56,130.35,129.70,129.60,129.50,128.96,128.72,128.52,127.75,126.47,125.92,125.75,119.39;HRMS(EI)Calcd.for C21H15N:[M+],281.1207;Found:281.1204。
其中,所述的1,4-二苯基-2,3-二甲酸甲酯基吡咯化学结构通
式为:
实施例2
一种1-邻甲苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(107mg)的邻甲基苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4b)289.7mg,产率83%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.43-7.46(m,2H),7.35-7.39(m,3H),7.26-7.32(m,3H),7.21-7.24(m,1H),6.92(s,1H),3.87(s,3H),3.67(s,3H),2.11(s,3H),;13C NMR(100MHz,CDCl3)δppm:167.1,150.0,139.0,135.4,133.2,130.6,129.1,128.7,127.5,127.3,127.2,126.5,125.8,124.7,123.0,121.6,52.5,51.9,17.4;HRMS(ESI-TOF)m/z calcd for C21H20NO4[M+H]+350.1387,found 350.1386。
其中,所述的1-邻甲苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例3
一种1-邻碘苯基-.4-二苯基-2,3-二甲酸甲酯基吡咯(4c)的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(219mg)的邻碘苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在110℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4c)309.5mg,产率67%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.92-7.94(m,1H),7.44-7.48(m,3H),7.36-7.40(m,3H),7.28-7.32(m,1H),6.93(s,1H),3.88(s,3H),3.69(s,3H);13C NMR(100MHz,CDCl3)δppm:167.0,159.7,142.8,139.3,133.0,130.4,128.9,128.7,128.3,127.5,127.2,125.6,125.0,122.4,122.3,97.5,52.6,52.0;HRMS(ESI-TOF)m/z calcd for C20H17NIO4[M+H]+462.0197,found 462.0207.
其中,所述的1-邻碘苯基-.4-二苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例4
一种1-间甲苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(107mg)的间甲基苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4d)294.2mg,产率84%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.43-7.46(m,2H),7.30-7.39(m,4H),7.14-7.19(m,3H),6.99(s,1H),3.84(s,3H),3.72(s,3H),2.41(s,3H);13C NMR(100MHz,CDCl3)δppm:166.8,160.5,139.4,139.0,133.2,129.3,128.71,128.69,128.59,128.17,127.7,127.1,126.6,125.9,123.2,110.5,52.4,52.0,21.3;HRMS(ESI-TOF)m/z calcd for C21H20NO4[M+H]+350.1387,found 350.1376.
其中,所述的1-间甲苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例5
一种1-间氟苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(111mg)的间氟苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在120℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4e)269.1mg,产率76%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.92-7.94(m,1H),7.44-7.48(m,3H),7.36-7.40(m,3H),7.28-7.32(m,1H),6.93(s,1H),3.88(s,3H),3.69(s,3H);13C NMR(100MHz,CDCl3)δppm:166.6,150.2,140.6,132.9,130.2,130.1,128.7,127.7,127.3,125.8,125.2,123.0,122.3,122.0,115.6,114.0,52.5,52.1;HRMS(ESI-TOF)m/z calcd for C20H17FNO4[M+H]+354.1136,found 354.1173.
其中,所述的1-间氟苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例6
一种1-间氯苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(127mg)的间氯苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4f)285.0mg,产率77%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.41-7.45(m,4H),7.36-7.40(m,4H),7.28-7.33(m,1H),7.00(s,1H),3.86(s,3H),3.74(s,3H);13C NMR(100MHz,CDCl3)δppm:166.6,160.1,140.1,134.5,132.8,129.9,128.8,128.7,127.6,127.3,126.5,125.9,125.2,124.6,122.9,122.4,52.5,52.0;HRMS(ESI-TOF)m/z calcd for C20H17ClNO4[M+H]+370.0841,found 370.0461.
其中,所述的1-间氯苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例7
一种1-间三氟苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(161mg)的间三氟苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在120℃条件下搅拌24小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4g)291.1mg,产率72%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.72(d,J=7.6Hz,1H),7.56-7.64(m,3H),7.44-7.46(m,2H),7.37-7.40(m,2H),7.30-7.34(m,1H),7.03(s,1H),3.88(s,3H),3.69(s,3H);13C NMR(100MHz,CDCl3)δppm:166.6,160.1,139.9,132.7,131.7,131.4,129.8,129.6,128.7,127.6,127.4,125.9,125.4,123.4,122.9,122.8,52.5,52.0;HRMS(ESI-TOF)m/z calcd for C21H17F3NO4[M+H]+404.1104,found 404.0739.
其中,所述的1-间三氟苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例8
一种1-对甲苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(107mg)的对甲基苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌15小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4h)329.1mg,产率94%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.43-7.45(m,2H),7.35-7.39(m,2H),7.29-7.31(m,1H),7.22-7.27(m,4H),6.98(s,1H),3.84(s,3H),3.72(s,3H),2.41(s,3H);13C NMR(100MHz,CDCl3)δppm:166.8,160.5,138.5,136.9,133.2,129.6,128.6,127.7,127.1,126.0,125.9,124.7,123.3,121.6,52.4,51.9,21.2;HRMS(ESI-TOF)m/z calcd for C21H20NO4[M+H]+350.1387,found 350.1419.
其中,所述的1-对甲苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例9
一种1-对甲氧苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(123mg)的对甲氧基苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌16小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4i)333.2mg,产率91%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.43-7.45(m,2H),7.35-7.39(m,2H),7.26-7.31(m,3H),6.95-6.97(m,2H),6.94(s,1H),3.85(s,6H),3.72(s,3H);13C NMR(100MHz,CDCl3)δppm:166.8,160.4,159.5,133.2,132.3,128.6,127.7,127.3,127.1,126.3,124.6,123.3,121.5,114.0,55.5,52.4,51.9;HRMS(ESI-TOF)m/z calcd for C21H20NO5[M+H]+366.1336,found 366.1343.
其中,所述的1-对甲氧苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例10
一种1-对氟苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(111mg)的対氟苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4j)276.2mg,产率78%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.42-7.45(m,2H),7.30-7.39(m,5H),7.11-7.17(m,2H),6.98(s,1H),3.85(s,3H),3.72(s,3H);13C NMR(100MHz,CDCl3)δppm:166.7,160.2,135.5,133.0,128.7,128.1,128.0,127.6,127.3,126.2,124.9,123.0,122.2,115.8,52.5,52.0;HRMS(ESI-TOF)m/z calcd for C20H17FNO4[M+H]+354.1136,found 354.1178.
其中,所述的1-对氟苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例11
一种1-对氯苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(93mg)的队氯苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4k)288.7mg,产率78%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.42-7.45(m,4H),7.36-7.41(m,2H),7.27-7.32(m,3H),6.98(s,1H),3.86(s,3H),3.73(s,3H);13C NMR(100MHz,CDCl3)δppm:166.7,160.2,137.9,134.5,132.9,129.2,128.7,127.60,127.55,127.3,126.0,124.4,122.4,110.0,52.5,52.0;HRMS(ESI-TOF)m/z calcd for C20H17ClNO4[M+H]+370.0841,found 370.0838.
其中,所述的1-对氯苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例12
一种1-对溴苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(171mg)的对溴苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4l)314.7mg,产率76%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.57-7.61(m,2H),7.36-7.44(m,4H),7.29-7.33(m,1H),7.22-7.26(m,2H),6.98(s,1H),3.86(s,3H),3.72(s,3H);13C NMR(100MHz,CDCl3)δppm:166.6,160.2,138.4,132.8,132.1,128.7,127.9,127.6,127.3,125.9,125.1,124.8,122.5,122.4,52.5,52.1;HRMS(ESI-TOF)m/z calcd for C20H17BrNO4[M+H]+414.0335,found 414.0313.
其中,所述的1-对溴苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例13
一种1-对三氟苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(161mg)的对三氟甲基苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在120℃条件下搅拌24小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4m)299.1mg,产率74%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.74(d,J=8.4Hz,2H),7.43-7.49(m,4H),7.36-7.40(t,2H),7.29–7.33(m,1H),7.02(s,1H),3.86(s,3H),3.74(s,3H);13C NMR(100MHz,CDCl3)δppm:166.5,160.1,142.3,132.7,130.6,128.7,127.6,127.4,126.7,126.2,125.8,125.4,122.9,117.0,55.6,52.1;HRMS(ESI-TOF)m/z calcd for C21H17FNO4[M+H]+404.1104,found 404.1103.
其中,所述的1-对三氟苯基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例14
一种1-(2,6-二甲基苯基)-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(121.1mg)的2,6-二甲基苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在110℃条件下搅拌22小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4n)265.9mg,产率73%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.46(d,J=7.6Hz,2H),7.36-7.39(m,2H),7.23-7.31(m,2H),7.14(d,J=7.6Hz,2H),6.85(s,1H),3.86(s,3H),3.67(s,3H),2.05(s,6H);13C NMR(100MHz,CDCl3)δppm:166.7,160.4,138.3,135.6,133.3,128.8,128.7,128.0,127.5,127.1,125.2,124.7,122.6,121.3,52.4,51.9,17.5;HRMS(ESI-TOF)m/z calcd for C22H22NO4[M+H]+364.1543,found 364.1547.
其中,所述的1-(2,6-二甲基苯基)-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例15
一种1-(2,4-二甲基苯基)-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(121.1mg)的2,4-二甲基苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4o)298.7mg,产率82%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.44-7.46(t,2H),7.36-7.39(m,2H),7.27-7.33(m,1H),7.01-7.12(m,3H),6.91(s,1H),3.87(s,3H),3.69(s,3H),2.38(s,3H),2.07(s,3H);13C NMR(100MHz,CDCl3)δppm:167.1,160.0,138.9,136.4,135.0,133.3,131.2,128.7,127.5,127.1,127.0,125.9,124.5,123.0.6,121.4,52.5,51.8,21.2,17.3;HRMS(ESI-TOF)m/z calcd for C22H22NO4[M+H]+364.1543,found 364.1589.
其中,所述的1-(2,4-二甲基苯基)-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例16
一种1-环己基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(99mg)的环己胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌17小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4p)297.6mg,产率87%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.32-7.40(m,4H),7.24-7.27(m,1H),7.08(s,1H),4.87-4.92(m,1H),3.84(s,3H),3.81(s,3H),2.14(d,J=11.2Hz,2H),1.90(d,J=12.8Hz,2H),1.43-1.64(m,4H),1.17-1.26(m,2H);13C NMR(100MHz,CDCl3)δppm:167.5,160.9,133.7,128.6,127.4,126.8,123.8,121.40,121.38,120.9,57.0,52.3,51.8,34.7,25.8,25.5;HRMS(ESI-TOF)m/z calcd for C20H24NO4[M+H]+342.1700,found 342.1700.
其中,所述的1-环己基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例17
一种1-苄基-4苯基-苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(107mg)的苄胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌19小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4q)308.1mg,产率88%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.31-7.39(m,7H),7.23-7.29(m,3H),7.16-7.18(m,2H),6.94(s,1H),5.52(s,2H),3.82(s,3H),3.78(s,3H);13C NMR(100MHz,CDCl3)δppm:167.3,160.7,137.1,133.3,128.9,128.7,127.9,127.4,127.2,127.0,125.8,124.0,122.3,121.2,52.5,51.9;HRMS(ESI-TOF)m/z calcd for C21H20NO4[M+H]+350.1387,found 350.1396.
其中,所述的1-苄基-4苯基-苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
实施例18
一种1,2,4-三苯基-3-甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(93mg)的苯胺,苯丙炔酸甲酯1.0mmol(160mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌16小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4r)336.5mg,产率95%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.50(d,J=7.2Hz,2H),7.35-7.41(m,3H),7.22-7.32(m,9H),7.09(d,J=6.8Hz,1H),6.96(s,1H),3.55(s,3H);13C NMR(100MHz,CDCl3)δppm:165.3,138.6,137.2,134.4,130.7,130.4,128.4,128.2,127.4,127.3,127.0,126.7,126.2,126.0,125.4,121.5,112.8,50.3;HRMS(ESI-TOF)m/z calcd for C24H20NO2[M+H]+354.1489,found 354.1488.
其中,所述的1,2,4-三苯基-3-甲酸甲酯基吡咯化学结构通式为:
实施例19
一种1.4-二苯基-3-甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(93mg)的苯胺,丙炔酸甲酯1.0mmol(84mg)在室温下搅拌10分钟,然后加入2.0mmol苯乙烯(208mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4s)189.1mg,产率68%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.77(d,J=2.8Hz,1H),7.54(d,J=7.6Hz,2H),7.43-7.49(m,4H),7.28-7.40(m,4H),7.07(d,J=6.4Hz,1H),3.76(s,3H);13C NMR(100MHz,CDCl3)δppm:164.9,139.6,134.3,129.9,129.3,128.5,127.9,120,126.9,126.1,120.8,119.8,115.1,51.1;HRMS(ESI-TOF)m/z calcd for C18H16NO2[M+H]+278.1176,found 278.1177.
其中,所述的1.4-二苯基-3-甲酸甲酯基吡咯化学结构通式为:
实施例20
一种1-苯基-4-对甲苯苯基-2,3-二甲酸甲酯基吡咯的合成方法,包括以下步骤:
在25mL耐压反应管中加入1.0mmol(93mg)的苯胺,丁炔二酸二甲酯1.0mmol(142mg)在室温下搅拌10分钟,然后加入2.0mmol对甲基苯乙烯(236mg),0.54g过二硫酸钾,11.2mg醋酸钯,2mL乙腈,2mL乙酸混合后,在100℃条件下搅拌20小时,反应后冷却至室温,加适量的饱和碳酸氢钠溶液,20mL二氯甲烷稀释,最后用10mL水洗,最后水相用10mL二氯甲烷洗两次,合并后的有机相我们用无水硫酸钠干燥。然后通过柱层析法提纯(乙酸乙酯:石油醚=20:1),最后的目标产物(4t)322.1mg,产率92%,纯度99.9%。1H NMR(400MHz,CDCl3)δppm:7.43-7.47(m,4H),7.33-7.36(m,4H),7.17-7.19(m,2H),6.99(s,1H),3.85(s,3H),3.71(s,3H),2.36(s,3H);13C NMR(100MHz,CDCl3)δppm:166.8,160.4,139.5,136.9,130.2,129.4,128.5,127.6,126.2,125.8,124.9,122.9,121.8,52.4,51.9,21.2;HRMS(ESI-TOF)m/z calcd for C21H20NO4[M+H]+350.1387,found 350.1385.
其中,所述的1-苯基-4-对甲苯苯基-2,3-二甲酸甲酯基吡咯化学结构通式为:
一种吡咯衍生物的合成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。








动态评分
0.0