

IPC分类号 : B01J21/18,B01J32/00,B01J23/50,B01J35/02,C07C215/76,C07C213/02

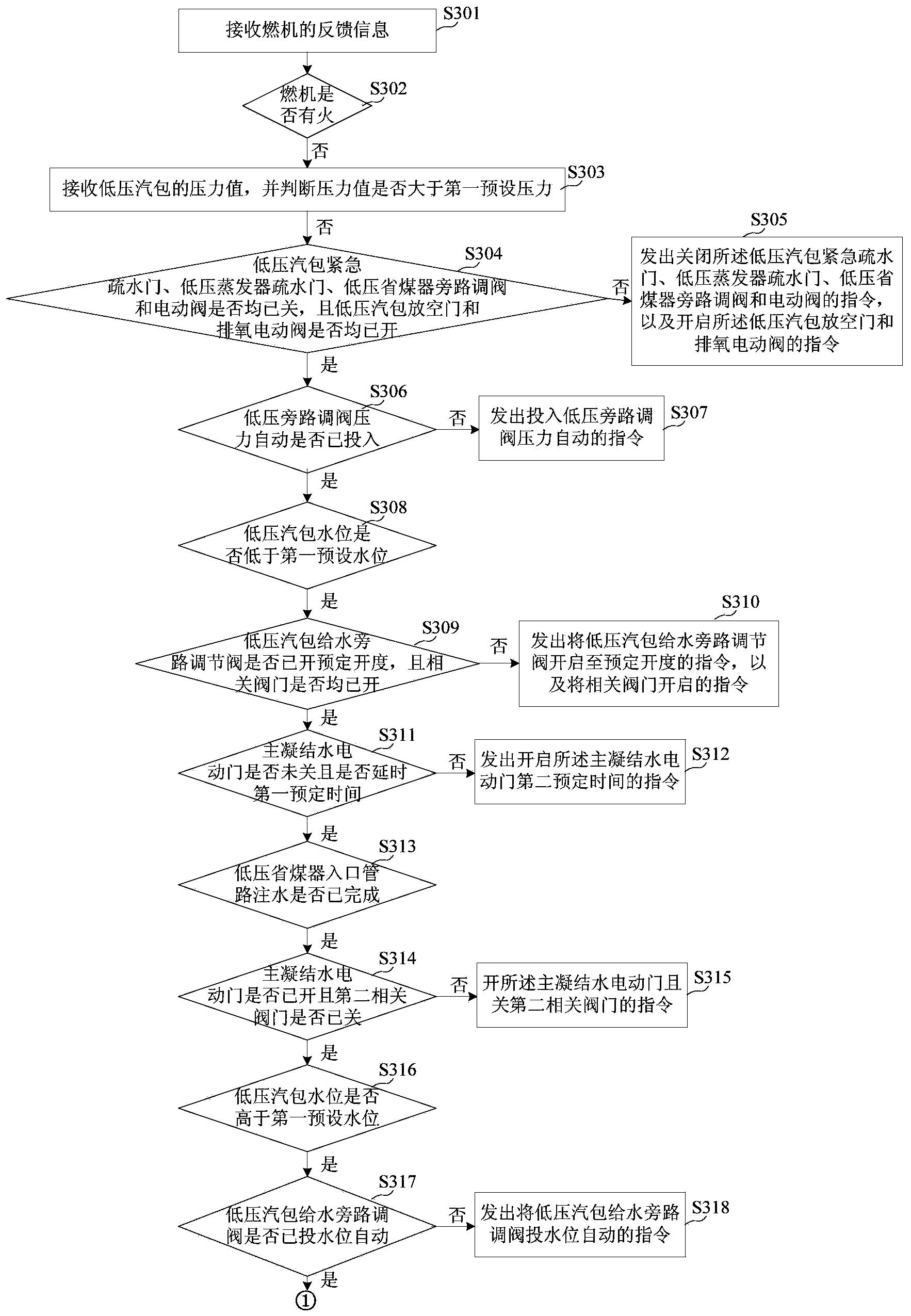






专利摘要
本发明涉及一种氧化石墨烯在非平面基底表面的自组装方法以及基于此方法而构筑的具有独特树莓状结构的二氧化硅/石墨烯/银纳米颗粒复合微球,属于石墨烯基复合材料制备的技术领域。先以正硅酸四乙酯为硅源,采用稍加改进的S?ber法制备二氧化硅微球,然后于其表面组装上阳离子聚电解质使其表面正电化,再在连续强力超声条件下通过静电作用使小尺寸氧化石墨烯片自组装于微球表面,接下来再在复合微球表面原位沉积银纳米颗粒,然后以水合肼将其中的氧化石墨烯还原成石墨烯,从而得到树莓状结构的二氧化硅/石墨烯/银纳米颗粒复合微球。该复合微球具有良好的水分散性,对4-硝基苯的催化还原表现出优异的催化活性,展现出了广阔的应用前景。
权利要求
1.一种在非平面基底表面自组装氧化石墨烯的方法,其特征在于自组装过程中不发生粘连和聚集行为,非平面基底为二氧化硅微球,其粒径范围为200~500 nm,且自组装过程中需要在功率高于150 W的连续强力超声条件下完成。
2.如权利要求1所述的氧化石墨烯,其特征在于其径向尺寸不超过200 nm。
3.如权利要求1所述的氧化石墨烯在二氧化硅微球表面的组装,其特征在于所组装的氧化石墨烯层厚度约为6 nm,且组装后形成的二氧化硅/氧化石墨烯复合微球具有极佳的水分散性,该复合微球经超声分散后形成的水分散液在静置过夜后仍无明显沉降或聚集体产生。
4.如权利要求1所述的氧化石墨烯在非平面基底表面自组装的具体步骤如下:
(1) 采用浓度为0.2 M的正硅酸四乙酯为硅源,将其与含有氨(浓度范围为0.8~2.5 M)和水(浓度为34 M)的乙醇溶液等体积混合,剧烈搅拌过夜,正硅酸四乙酯在氨的催化下于乙醇介质中水解得到二氧化硅胶体微球,所制备的二氧化硅胶体微球尺寸可以通过调控氨的浓度加以控制;
(2) 将步骤(1)制备的二氧化硅微球超声分散于水中,然后将其与过量的聚二烯丙基二甲基氯化铵水溶液混合,剧烈搅拌过夜,使阳离子聚电解质聚二烯丙基二甲基氯化铵通过静电作用组装于二氧化硅微球表面,得到表面正电化的二氧化硅微球;
(3) 将小尺寸氧化石墨烯片预先超声分散于水中,形成一定浓度的水分散液,将该水分散液在高速下离心5 min,除去其中的少量聚集体,并取上层分散液再次超声30 min,用于接下来的组装;
(4) 将步骤(2)制备的表面正电化的二氧化硅微球超声分散于水中,将其在连续强力超声条件下逐滴加入至过量的步骤(3)中的小尺寸氧化石墨烯水分散液中,接着超声30 min以完成小尺寸氧化石墨烯片在表面正电化的二氧化硅微球表面的组装;然后将反应体系在一定转速下离心5 min使二氧化硅/氧化石墨烯复合微球与未组装上的游离氧化石墨烯水分散液得以分离。
5.一种具有独特树莓状结构的二氧化硅/石墨烯/银纳米颗粒复合微球,其特征在于银纳米颗粒均匀分布于复合微球表面,其尺寸范围为2~50 nm,该复合微球具有极好的水分散性,经超声分散后形成的水分散液在静置过夜后仍无明显沉降或聚集体产生,且对4-硝基的催化还原表现出优异的催化活性。
6.如权利要求5所述的二氧化硅/石墨烯/银纳米颗粒复合微球的制备方法,其特征在于具体合成步骤如下:
(1) 将权利要求3或4中的二氧化硅/氧化石墨烯复合微球超声分散于水中,将其与过量的新制银氨溶液搅拌混合,于85 oC下反应45 min,利用氧化石墨烯本身的还原性将银氨络合物原位还原成银纳米颗粒并沉积于复合微球表面,从而得到二氧化硅/氧化石墨烯/银纳米颗粒复合微球;
(2) 将步骤(1)中的二氧化硅/氧化石墨烯/银纳米颗粒复合微球超声分散于水中,然后搅拌条件下逐滴加入过量水合肼,再于85 oC下反应1 h,将其中的氧化石墨烯成分将还原成石墨烯,即得到二氧化硅/石墨烯/银纳米颗粒复合微球。
说明书
技术领域
本发明属于石墨烯基复合材料制备的技术领域,具体涉及一种氧化石墨烯在超声辅助下于非平面基底(二氧化硅微球)表面自组装的方法以及基于此方法而构筑的具有高水分散性和高催化性能的树莓状结构二氧化硅/石墨烯/银纳米颗粒复合微球。
背景技术
石墨烯由于其独特的结构和优异的物理化学性能使得其成为了材料研究领域的热点,各种基于石墨烯的复合材料和功能材料也层出不穷。在以往的报道中,人们常将各种纳米颗粒(如金属氧化物、金属硫化物、贵金属、导电聚合物纳米颗粒等)负载于尺寸较大的石墨烯片(径向尺寸范围约为500 nm~5 μm)的表面来制备石墨烯/纳米颗粒复合材料。在制备这些石墨烯/纳米颗粒复合材料过程中,纳米颗粒通常是作为客体物质沉积或生长于作为主体物质的石墨烯表面,石墨烯充当了基底或支撑材料的角色,采取这种策略一方面容易引发聚集行为,另一方面也不利于制备形貌更为复杂、性能更为出色的石墨烯基复合材料。相反,通过在基底材料表面,尤其是非平面基底表面引入氧化石墨烯或石墨烯来构筑石墨烯基复合材料的研究则相对较少。最近,人们开始关注这一问题,并试图在各种微球如二氧化钛、四氧化三钴、硫化镉微球表面包覆一层氧化石墨烯并以此为基础来制备石墨烯基复合材料[(a) Lee, J. S.; You, K. H.; Park, C. B. Adv. Mater. 2012, 24, 1084—1088. (b) Yang, S.; Feng, X.; Ivanovici, S.; Müllen, K. Angew. Chem. Int. Ed. 2010, 49, 8404—8411. (c) Chen, Z.; Liu, S.; Yang, M.-Q.; Xu, Y.-J. ACS Appl. Mater. Interfaces 2013, 5, 4309—4319.]。然而在包覆过程中微球之间往往会被氧化石墨烯所粘连,产生严重的聚集现象,导致最终所制备的石墨烯基复合材料的水分散性很差,极大地限制了材料的各种性能和进一步功能化修饰。因此,如何克服氧化石墨烯在非平面基底表面组装时易产生的粘连和聚集现象,继而制备出结构独特、水分散性好、性能优异的石墨烯基复合材料成为了当前有关石墨烯研究的重点和难点。
发明内容
本发明目的之一是提供一种方便、简单的于非平面基底(二氧化硅微球)表面自组装氧化石墨烯的方法用以克服氧化石墨烯在基底表面自组装过程中常见的粘连和聚集现象;目的之二是在该方法的基础之上进一步制备出具有高水分散性、高催化活性和独特树莓状结构的二氧化硅/石墨烯/银纳米颗粒复合微球用以实现4-硝基苯酚高选择性的催化还原。
本发明是通过以下技术方案实现的,首先制备二氧化硅胶体微球,在其表面通过静电作用组装上阳离子聚电解质聚二烯丙基二甲基氯化铵,使其表面正电化,再在连续强力超声条件下通过静电作用实现小尺寸氧化石墨烯片在微球表面的组装,然后利用该氧化石墨烯包覆层本身的还原性将新制备的银氨络合物原位还原成银纳米颗粒并沉积于其表面,再以水合肼将所沉积的氧化石墨烯成分还原成石墨烯,从而得到具有独特树莓状结构并具有高水分散性的二氧化硅/石墨烯/银纳米颗粒复合微球,然后将该复合微球应用于4-硝基苯酚的催化还原,具体包括以下步骤:
(1) 采用浓度为0.2 M的正硅酸四乙酯为硅源,将其与含有氨(浓度范围为0.8~2.5 M)和水(浓度为34 M)的乙醇溶液等体积混合,剧烈搅拌过夜。正硅酸四乙酯在氨的催化下于乙醇介质中水解得到二氧化硅胶体微球。所制备的二氧化硅胶体微球尺寸可以通过调控氨的浓度加以控制。
(2) 将步骤(1)制备的二氧化硅微球超声分散于水中,然后将其与过量的聚二烯丙基二甲基氯化铵水溶液混合,剧烈搅拌过夜,使阳离子聚电解质聚二烯丙基二甲基氯化铵通过静电作用组装于二氧化硅微球表面,得到表面正电化的二氧化硅微球。
(3) 将小尺寸氧化石墨烯片预先超声分散于水中,形成一定浓度的水分散液,将该水分散液在高速下离心5 min,除去其中的少量聚集体,并取上层分散液再次超声30 min,用于接下来的组装。
(4) 将步骤(2)制备的表面正电化的二氧化硅微球超声分散于水中,将其在连续强力超声条件下逐滴加入至过量的步骤(3)中的小尺寸氧化石墨烯水分散液中,接着超声30 min以完成小尺寸氧化石墨烯片在表面正电化的二氧化硅微球表面的组装。然后将反应体系在一定转速下离心5 min使二氧化硅/氧化石墨烯复合微球与未组装上的游离氧化石墨烯水分散液得以分离。
(5) 将步骤(4)中的二氧化硅/氧化石墨烯复合微球超声分散于水中,将其与过量的新制银氨溶液搅拌混合,于85 oC下反应45 min,利用氧化石墨烯本身的还原性将银氨络合物原位还原成银纳米颗粒并沉积于复合微球表面,从而得到二氧化硅/氧化石墨烯/银纳米颗粒复合微球。
(6) 将步骤(5)中的二氧化硅/氧化石墨烯/银纳米颗粒复合微球超声分散于水中,然后搅拌条件下逐滴加入过量水合肼,再于85 oC下反应1 h,将其中的氧化石墨烯成分将还原成石墨烯,从而得到二氧化硅/石墨烯/银纳米颗粒复合微球。
(7) 将步骤(6)中的二氧化硅/石墨烯/银纳米颗粒复合微球超声分散于水中,再稀释至一定浓度,取其中的1 mL,将其依次与1 mL的硼氢化钠水溶液以及1 mL的4-硝基苯酚水溶液充分混合,然后将此反应混合物转移至比色皿中反应。该反应过程采用紫外—可见光谱原位监测,以测定该反应的速率常数并鉴定所制备的催化剂(即二氧化硅/石墨烯/银纳米颗粒复合微球)的催化活性。
本发明所述二氧化硅微球尺寸十分均一,其粒径随步骤(1)中催化剂氨浓度的增加而增大,其尺寸可调范围为200~500 nm,并且均具有良好的水分散性。
本发明所述小尺寸氧化石墨烯片,其径向尺寸均不超过200 nm。为避免氧化石墨烯在二氧化硅微球表面自组装过程中易发生的粘连和聚集现象,小尺寸氧化石墨烯水分散液在用于组装前需在高速(转速高于15000 rpm)下离心5 min,以除去其中少量的聚集体。
本发明所述超声均为连续强力超声,其功率高于150W。
与现有技术相比,本发明具有如下优点和效果:
1、本发明通过采用小尺寸氧化石墨烯片在连续强力超声条件下于二氧化硅微球表面进行组装,有效解决了氧化石墨烯与微球复合过程中易发生的粘连和聚集现象,为实现氧化石墨烯在其它非平面基底表面的组装提供了行之有效的方法。
2、本发明中所制备的氧化石墨烯基和石墨烯基复合微球(如二氧化硅/氧化石墨烯、二氧化硅/氧化石墨烯/银纳米颗粒、二氧化硅/石墨烯/银纳米颗粒复合微球)均具有良好的水分散性,将其真空干燥后可重新超声分散于水中,且静置过夜后也不发生明显沉降或产生大块聚集体,因而具有良好的应用潜能。
3、本发明制备的二氧化硅/石墨烯/银纳米颗粒复合微球具有独特的树莓状结构,其良好的水分散性以及银纳米颗粒与石墨烯之间的协同作用使其催化性能远远强于其它已经报道的含银纳米颗粒的同类催化剂。
4、本发明中的所有制备和反应过程均以水或乙醇作为介质,而且反应条件温,操作方法简便,成本低廉,污染极小。此外,所制备的目标材料二氧化硅/石墨烯/银纳米颗粒复合微球也具有持久的化学稳定性和催化活性。
附图说明
图1 是二氧化硅/石墨烯/银纳米颗粒复合微球的制备示意图。
图2 是不同倍率下二氧化硅微球的SEM图。
图3是小尺寸氧化石墨烯片的AFM图。
图4是不同倍率下二氧化硅/氧化石墨烯复合微球的SEM图。
图5是不同倍率下二氧化硅/氧化石墨烯复合微球的TEM图。
图6是二氧化硅/氧化石墨烯复合微球水分散液的紫外—可见光谱图。
图7是二氧化硅/氧化石墨烯复合微球水分散液静置过夜后的电子照片。
图8 是不同倍率下二氧化硅/氧化石墨烯/银纳米颗粒复合微球的SEM图。
图9 是不同倍率下二氧化硅/石墨烯/银纳米颗粒复合微球的SEM图。
图10是不同倍率下二氧化硅/石墨烯/银纳米颗粒复合微球的TEM图。
图11 是二氧化硅/石墨烯/银纳米颗粒复合微球表面银纳米颗粒的高分辨TEM图。
图12 是二氧化硅/石墨烯/银纳米颗粒复合微球水分散液的紫外—可见光谱图。
图13是二氧化硅/石墨烯/银纳米颗粒复合微球水分散液静置过夜后的电子照片。
图14是二氧化硅/石墨烯/银纳米颗粒复合微球催化硼氢化钠还原4-硝基苯酚效果图。
具体实施方式
下面通过实施例并结合附图对本发明作进一步具体说明。
实施例1:将本发明提供的方法用于小尺寸氧化石墨烯片在二氧化硅微球表面的组装:
(1) 将100 mL浓度为0.2 M的正硅酸四乙酯乙醇溶液与100 mL含有氨(浓度为1.6 M)和水(浓度为34 M)的乙醇溶液混合,剧烈搅拌过夜。采用乙醇为溶剂,将正硅酸四乙酯在氨催化下水解生成的二氧化硅胶体微球反复离心—洗涤后真空干燥。所制备的二氧化硅胶体微球的尺寸十分均一,其平均粒径 ~ 280 nm (图2)。
(2) 将1 g步骤(1)制备的二氧化硅微球超声分散于100 mL水中,配置成10 mg/mL的水分散液;再将5 g质量分数为20%的的聚二烯丙基二甲基氯化铵溶于95 mL水中,并搅拌1 h以上。然后将二氧化硅水分散液与聚二烯丙基二甲基氯化铵水溶液混合,剧烈搅拌过夜,使阳离子聚电解质聚二烯丙基二甲基氯化铵通过静电作用组装于二氧化硅微球表面。采用水为溶剂,将所得到的该表面正电化的二氧化硅微球反复离心—洗涤后真空干燥。
(3) 将小尺寸氧化石墨烯片预先分散于水中,超声60 min,形成0.2 mg/mL的水分散液,将该水分散液在16000 rpm的转速下离心5 min,除去其中的少量聚集体,并取上层分散液再次超声30 min,所得小尺寸氧化石墨烯水分散液用于接下来的组装,其径向尺寸均不超过200 nm (图3)。
(4) 将0.8 g步骤(2)制备的表面正电化的二氧化硅微球超声分散于100 mL水中,形成浓度为8 mg/mL的水分散液,将其在200 W的连续强力超声条件下逐滴加入至100 mL步骤(3)中的小尺寸氧化石墨烯水分散液中,接着超声30 min以完成小尺寸氧化石墨烯片在表面正电化二氧化硅微球表面的组装,从而得到二氧化硅/氧化石墨烯复合微球。然后将反应体系在8000 rpm的转速下离心5 min,使二氧化硅/氧化石墨烯复合微球与未组装的游离氧化石墨烯水分散液分离,并以水为溶剂将二氧化硅/氧化石墨烯复合微球超声分散洗涤,并再次离心分离后真空干燥。
(5) 称取9 mg步骤(4)制备的二氧化硅/氧化石墨烯复合微球,将其超声分散于3 mL水中,形成浓度为3 mg/mL的水分散液,静置过夜后观察沉降情况。
所制备的二氧化硅/氧化石墨烯复合微球之间不被氧化石墨烯所粘连(图4和图5),而且二氧化硅微球表面所组装包覆的氧化石墨烯层十分均匀,平均厚度 ~ 6 nm (图5),其组装量占二氧化硅/氧化石墨烯复合微球质量的1.45%。
所制备的二氧化硅/氧化石墨烯复合微球水分散液在230 nm处具有一吸收峰(图6),为氧化石墨烯的特征吸收峰,这进一步证实了氧化石墨烯在二氧化硅微球表面的成功组装。
所制备的二氧化硅/氧化石墨烯复合微球水分散性极佳,干燥后的二氧化硅/氧化石墨烯复合微球经超声可再次分散于水中,形成均一的棕色水分散液,并且静置过夜后也不产生明显沉降或大块聚集体(图7)。
实施例2:将本发明提供的方法用于制备具有独特结构和高水分散性的石墨烯基复合材料:
(1) 将0.4 g实施例1中的二氧化硅/氧化石墨烯复合微球超声分散于100 mL水中,形成浓度为4 mg/mL的水分散液,将其与100 mL浓度为40 mM的新制银氨溶液搅拌混合,于85 oC下反应45 min,利用氧化石墨烯本身的还原性将银氨络合物原位还原成银纳米颗粒并沉积于复合微球表面,从而制备得到二氧化硅/氧化石墨烯/银纳米颗粒复合微球。采用水为溶剂,将其反复离心—洗涤后真空干燥。
(2) 将0.2 g步骤(1)中的二氧化硅/氧化石墨烯/银纳米颗粒复合微球超声分散于100 mL水中,形成浓度为2 mg/mL的水分散液,然后搅拌条件下逐滴加入水合肼,使其最终浓度达到10 mg/mL,再于85 oC下反应1 h,将其中的氧化石墨烯成分将还原成石墨烯,从而得到二氧化硅/石墨烯/银纳米颗粒复合微球。采用水为溶剂,将其反复离心—洗涤后真空干燥。
(3) 称取0.9 mg步骤(2)制备的二氧化硅/石墨烯/银纳米颗粒复合微球,将其超声分散于3 mL水中,形成浓度为0.3 mg/mL的水分散液,静置过夜后观察沉降情况。
所制备的二氧化硅/石墨烯/银纳米颗粒复合微球之间也无粘连,并且具有独特的树莓状结构(图9和图10),其形貌与二氧化硅/氧化石墨烯/银纳米颗粒复合微球无明显差异(图8),说明经水合肼热处理后形貌得仍然以保持。
所制备的二氧化硅/石墨烯/银纳米颗粒复合微球表面均匀分布着银纳米颗粒,其粒径范围为2~50 nm (图10),而且其晶格条纹清晰可见,其0.236 nm的晶格间距很好地对应于银纳米颗粒的(111)晶面(图11),这很好地说明银纳米颗粒在复合微球表面的成功沉积。
所制备的二氧化硅/石墨烯/银纳米颗粒复合微球水分散液在260 nm处具有一吸收峰(图12),为石墨烯的特征吸收峰,这说明二氧化硅/氧化石墨烯/银纳米颗粒复合微球经水合肼热处理后,其中所沉积的氧化石墨烯成分确实被还原成了石墨烯;同时,所制备的二氧化硅/石墨烯/银纳米颗粒复合微球水分散液在410 nm处还具有另一吸收峰(图12),很好地对应了所沉积银纳米颗粒典型的表面等离子体共振吸收峰。此外,所制备的二氧化硅/石墨烯/银纳米颗粒复合微球中银含量为4%。
所制备的二氧化硅/石墨烯/银纳米颗粒复合微球也具有极好的水分散性,干燥后的二氧化硅/石墨烯/银纳米颗粒复合微球经超声可再次分散于水中,形成均一的红色水分散液,并且静置过夜后也不产生明显沉降或大块聚集体(图13)。
实施例3:将本发明中制备的石墨烯基复合材用于4-硝基苯酚的催化还原:
(1) 称取3 mg实施例2中的二氧化硅/石墨烯/银纳米颗粒复合微球,将其超声分散于20 mL水中,形成浓度为0.15 mg/mL的水分散液,取其中1 mL,然后将其稀释成浓度为1.5 μg/mL的水分散液。
(2) 取步骤(1)中稀释后的水分散液的1 mL,将其依次与1 mL浓度为30 mM的硼氢化钠水溶液以及1 mL浓度为0.3 mM的4-硝基苯酚水溶液充分混合,然后将此反应混合物转移至比色皿中反应。
(3) 该催化反应整个过程采用紫外—可见光谱进行原位监测,以测定该反应的速率常数并鉴定所制备的催化剂(即二氧化硅/石墨烯/银纳米颗粒复合微球)的催化活性。
图14为二氧化硅/石墨烯/银纳米颗粒复合微球催化硼氢化钠还原4-硝基苯酚效果图,从中可以看到步骤(2)中所述反应体系的紫外—可见光谱随时间的变化。很明显,该催化反应在9 min之内就可以完成,且转化率超过99.5%;此外,该反应近似于一级反应,其速率常数为0.7 min?1。由此可见,所制备的二氧化硅/石墨烯/银纳米颗粒复合微球具有十分出色的催化性能,远高于含银纳米颗粒的同类催化剂,说明其具有良好的应用前景。
一种基于氧化石墨烯自组装制备石墨烯基复合材料的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0