

IPC分类号 : C08G65/00,C08G65/48,C08G63/91,C07D285/00,C07D417/12,C07C323/60,C07C319/20,C07C323/42,C07D249/04,A61K31/337,A61K31/015,A61K31/4745,A61K47/34,A61K47/22,A61K47/20










专利摘要
本发明公开了通过分子胶连接的两亲性嵌段共聚物及其合成方法与应用,还公开了合成该共聚物的亲水化合物、疏水化合物及其制备方法。该共聚物M结构式如下式所示: 本发明是通过分别取亲水化合物、疏水化合物和氧化剂,在卤代烃溶剂中进行混合,制得所述两亲性嵌段共聚物M。该共聚物M可作为药物运输载体;所述药物为难溶于水的药物。本发明化合物合成原料简单易得,合成过程均为常规反应,反应条件温和,可以大量制备;由该类嵌段共聚物自组装而成的胶束可用于药物运输载体。
说明书
技术领域
本发明涉及化学合成和生物医药领域,尤其涉及一类新型的通过分子胶连接的两亲性嵌段共聚物及其合成方法与应用。
背景技术
两亲性嵌段共聚物胶束由具有亲水性片段和疏水性片段组成,在水溶液中自发形成的一种自组装结构,具有粒径小、粒度分布窄、结构稳定、载药范围广、体内滞留时间长、载药量高和独特的体内分布等特点。
嵌段共聚物胶束能对难溶性药物有效增溶,可作为难溶性抗肿瘤药、降压药、抗菌药、基因治疗药等药物载体,已受到广泛关注。用其作为抗肿瘤药物载体不仅能提高疗效、降低毒副作用,还能有效改善抗肿瘤药物的多药耐药性(MDR)。因此嵌段共聚物胶束作为难溶性抗肿瘤药物载体具有广阔的发展前景。
基于这一思路,近年来有关嵌段高聚物载体材料的研究非常热,如二嵌段AB高聚物,三嵌段ABA高聚物,以及多臂型、星型AnBm高聚物等。特别是具有两亲性特性的高聚物,在一定条件下,在溶液中自组装成为胶束结构的纳米粒子,中间是疏水的核,周围是亲水的壳,难溶性化合物如果能够稳定地分布在疏水核中,就可以被有效增溶。除胶束结构外,两亲性嵌段聚合物还可以用来稳定固体纳米粒子和乳剂等等,由于其结构明确,纯度高,一般具有比小分子表面活性剂低的临界胶束浓度,所以体内应用毒性较小,被认为是药物纳米制剂较为理想的材料之一。
目前传统的纳米制剂的合成和筛选往往脱离具体药物的结构,具有盲目性。其合成方法繁琐,很难获得系统的组合材料库以备筛选。所以尽管有大量的研究,但真正有临床应用价值的只有PLA和PEG。理想的情况应该是针对特定的一个或一类化合物结构,为其量身订做制剂材料,并对材料的结构、组成、分子量等进行系统的筛选和优化,才能最有效的建立稳定的药物分子分散体系。
发明内容
本发明的目的在于基于聚乙二醇的亲水性和聚乳酸的疏水性的思路,提供了一类新型的通过分子胶连接的两亲性嵌段共聚物及其合成方法与应用。由该类嵌段共聚物自组装而成的胶束可用于药物运输载体。
本发明的目的是通过以下技术方案来实现的:
第一方面,本发明涉及一种两亲性嵌段共聚物M,其结构式如式(I)所示:
其中,n为1~300中的任意一个整数;
R为烷基链,
或为式(II)所示的结构:
其中m为1~100的任意一个整数,
或为式(III)所示的结构:
其中k为1~100中的任意一个整数。
第二方面,本发明涉及一种制备如上述的两亲性嵌段共聚物M的方法,包括如下步骤:分别制备亲水化合物和疏水化合物;之后取亲水化合物、疏水化合物和氧化剂在卤代烃溶剂中进行混合,即得所述两亲性嵌段共聚物M。
优选的,所述步骤c具体为:分别取亲水化合物和疏水化合物的二氯甲烷溶液,混合均匀旋除二氯甲烷,残留物用I2的二氯甲烷溶液溶解、反应;其中,所述的亲水化合物、疏水化合物和I2的摩尔比为1∶1~10∶1~20。
第三方面,本发明涉及一种亲水化合物E4,其结构式如式(IV)所示:
其中,n为1~300中的任意一个整数。该亲水化合物用于制备前述的两亲性嵌段共聚物M。
第四方面,本发明涉及一种制备上述的亲水化合物E4的方法,包括如下步骤:
a、E2的合成,在碱性条件下,取3,5-二硝基苯甲酰氯(E1)与末端为氨基的PEG进行酰化反应,得化合物E2;
b、E3的合成,用氢气将步骤a制得的化合物E2中的硝基还原为氨基,得化合物E3;
c、E4的合成,在缩合剂作用下,将步骤b制得的化合物E3与酸反应,得所述亲水化合物E4。
优选的,
所述步骤a具体为:以DCM(二氯甲烷)做溶剂,PEG(聚乙二醇)在NEt3(三乙胺)的作用下,和E1在0℃下反应,TLC跟踪至反应完全;反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得红褐色液体产品;其中,所述的PEG、NEt3和E1的摩尔比为1∶1.4~1.5∶1.5~4;
所述步骤b具体为:以甲醇做溶剂,E2在Pd/C(其中,Pd的质量百分比含量为10%)和H2的作用下反应,温度为30℃,TLC跟踪至反应完全;反应结束后,硅藻土滤除Pd/C,得红褐色液体产品;其中,所述的E2和Pd/C的质量比为1∶0.15~0.2;
所述步骤c具体为:以DCM做溶剂,控制温度0℃,先用EDCl (1-(3-二甲氨基丙基)-3-乙基碳二亚胺)、HOBt(1-羟基苯并三唑)把(4-(三苯巯基)丁酸)S3活化完全,以活化后的物质和E3在30℃下作用;TLC跟踪至反应完全,反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得红褐色液体产品;其中,所述的EDCl、HOBt、S3和E3的摩尔比为3.6∶3.6∶3~5∶1。
第五方面,本发明涉及一种疏水化合物P1,其结构式如式(V)所示:
其中,R为烷基链。该疏水化合物用于制备前述的两亲性嵌段共聚物M。
第六方面,本发明涉及一种制备上述的疏水化合物P1的方法,包括如下步骤:
a、Z2的合成,在缩合剂作用下,Z1(5-氨基间苯二甲酸)上的羧基和氨基反应得化合物Z2;
b、P1的合成,在碱性条件下,步骤a制得的化合物Z2上的氨基与酰氯反应得所述疏水化合物P1。
优选的,
所述步骤a具体为:以DMF(N,N-二甲基甲酰胺)做溶剂,控制温度0℃,先用EDCl、HOBt把Z1活化完全,以活化后的物质和(2-(三苯巯基)乙胺)N2,在30℃下作用;TLC跟踪至反应完全,反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色泡沫状固体产品;其中,所述的EDCl、HOBt、N2和Z1的摩尔比为2.4∶2.4∶3∶1;
所述步骤b具体为:以DCM做溶剂,Z2在NEt3的作用下,和烷基酰氯在0℃下反应,TLC跟踪至反应完全;反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色泡沫状固体产品;其中,所述的Z2、NEt3和烷基酰氯的摩尔比为1∶3∶2。
第七方面,本发明涉及一种疏水化合物P2,其结构式如式(VI)所示:
其中,m为1~100中的任一整数。该疏水化合物用于制备前述的两亲性嵌段共聚物M。
第八方面,本发明涉及一种制备上述的疏水化合物P2的方法,包括如下步骤:
第一步,制备化合物Z4,具体如下:
a、Z3的合成,化合物Z2与丁二酸酐在有机溶剂中反应得化合物Z3;
b、Z4的合成,在缩合剂作用下,步骤b中制得的化合物Z3与丙炔胺反应得化合物Z4;
第二步,制备化合物A3,具体如下:
a、A2的合成,A1与氯乙酰氯反应得化合物A2,所述A1的结构式如式(VII)所示:
其中,m为1~100的任一整数;
b、A3的合成,步骤a制得的化合物A2与叠氮钠反应得化合物A3;
第三步,将第一步制得的Z4与第二步制得的A3反应,制备得到所述疏水化合物P2。
优选的,
第一步中,步骤a具体为:以CHCl3做溶剂,Z2和丁二酸酐在常温下反应,TLC跟踪至反应完全;反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色泡沫状固体产品;其中,所述的Z2和丁二酸酐的摩尔比为1∶1.5~2;
步骤b具体为:以DCM做溶剂,控制温度0℃,先用EDCl、HOBt把Z3活化完全,以活化后的物质和丙炔胺在30℃反应;TLC跟踪至反应完全,反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色泡沫状固体产品;其中,所述的EDCl、HOBt、Z3和丙炔胺的摩尔比为1.2∶1.2∶1∶4;
第二步中,所述步骤a具体为:以DCM做溶剂,A1在NEt3的作用下,和氯乙酰氯在0℃下反应,TLC跟踪至反应完全;反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得淡黄色油状液体产品;其中,所述的A1、NEt3和氯乙酰氯的摩尔比为1∶6∶10;
步骤b具体为:以DMSO做溶剂,A2和NaN3在常温下反应,TLC跟踪至反应完全;反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得淡黄色油状液体产品;其中,所述的A2和NaN3的摩尔比为1∶5~10;
第三步具体为:将Z4和A3溶于适量THF,体系多次抽充氮气,加氮气球,使反应在氮气保护下进行;将无水CuSO4固体和VcNa(抗坏血酸钠)混合,抽真空,加入去离子水震荡得黄色悬浊液,再注入反应体系中,室温反应;反应结束后,加入适量DCM萃取体系,水洗后无水硫酸钠干燥,柱色谱分离得白色泡沫状固体;其中,所述的Z4、A3、CuSO4和VcNa的摩尔比为1∶1∶0.6∶2~3。
第九方面,本发明涉及一种疏水化合物P3,其结构式如式(VIII)所示:
其中k为1~100中的任意一个整数。该疏水化合物用于制备前述的两亲性嵌段共聚物M。
第十方面,本发明涉及一种制备上述的疏水化合物P3的方法,包括如下步骤:
a、B2的合成,B1与丁二酸酐反应得B2,所述B1的结构式如式(IX)所示:
其中,k为1~100中的任意一个整数;
b、P3的合成,在缩合剂作用下,步骤a制得的化合物B2与Z2反应得P3。
优选的,
所述步骤a具体为:以DMF做溶剂,B1在DMAP的作用下,和丁二酸酐在室温反应;TLC跟踪至反应完全;反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色粘稠的液体产品;其中,所述的B1、DMAP和丁二酸酐的摩尔比为1∶5∶1~2;
所述步骤b具体为:以DMF做溶剂,控制温度0℃,先用EDCl、HOBt把B2活化完全,以活化后的物质和Z2在30℃反应;TLC跟踪至反应完全,反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色泡沫状固体产品;其中,所述的EDCl、HOBt、B3和Z2的摩尔比为3.6∶3.6∶3∶1。
第十一方面,一种如前述的两亲性嵌段共聚物M在制备药物运输载体中的用途。
优选的,所述药物为难溶于水的药物。用这种载体包裹后,可有效地改善药物的水溶性。
优选的,所述难溶于水的药物为紫杉醇、榄香烯或喜树碱。
本发明具有如下有益效果:
1、该类化合物合成原料简单易得,合成过程均为常规反应,反应条件温和,可以大量制备。
2、该类化合物可在一定条件下形成胶束,由该类嵌段共聚物自组装而成的胶束可用于药物运输载体。
附图说明
图1为亲水化合物E4的合成路径图;
图2为疏水化合物P1的合成路径图;
图3为疏水化合物P2的合成路径图;
图4为疏水化合物P3的合成路径图;
图5为两亲性嵌段共聚物M的合成路径图;
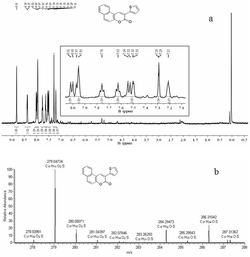
图6为A2和A3的红外谱图,其中a为A3,b为A2;
图7为A2和A3的红外谱图,其中a为A3,b为A2;
图8为E4、P1和M1的红外谱图,其中a为E4,b为P1,c为M1;
图9为PEG、E4和M1的GPC图,其中a为E4,b为PEG,c为M1;
图10为E4、P2和M2的红外谱图,其中a为E4,b为P2,c为M2;
图11为PEG、E4和M2的GPC图,其中a为PEG,b为E4,c为M2;
图12为E4、P2和M3的红外谱图,其中a为E4,b为P3,c为M3;
图13为PEG、E4和M3的GPC图,其中a为E4,b为PEG,c为M3;
图14为P3、E4和M4的红外谱图,其中a为P3,b为E4,c为M4;
图15为E4、P3和M4的GPC图,其中a为E4,b为P3,c为M4;
其中,图6、7、8、10、12、14、15中的纵坐标为透过率(transmittance%),横坐标为波长(wavelength)CM-1。
具体实施方式
下面结合附图和具体实施例对本发明进一步进行阐述,但本发明的保护范围不限于下述的实施例。
以下实施例中所用的原料、试剂均为市售AR、CP级。本发明所得中间产物及最终产物采用NMR,IR,GPC等进行表征。
实施例1、亲水化合物E4的合成
所述亲水化合物E4,其结构式如下式所示:
其中,n为1~300中的任意一个整数。
1.1当n=3时,亲水化合物E4的合成
其合成路径图如图1所示:
(1)E2的合成,在碱性条件下,取化合物E1与末端为氨基的PEG进行酰化反应,得化合物E2;
所述步骤具体为:在一单口烧瓶中加入100ml二氯甲烷,而后依次加入化合物末端为氨基的PEG164(12.0g,16.0mmol),三乙胺(3.34ml,24.0mmol),E1(5.53g,24.0mmol)控制0℃下搅拌10min。室温(25℃)下反应30min。停止反应以水洗涤,无水硫酸钠干燥,得红褐色液体15.37g。柱色谱分离,产率94.0%。
1H NMR(CDCl3,300MHz)δ9.23(t,J=2.0Hz,1H,ArH),9.19(d,J=2.0Hz,2H,ArH),4.60(t,J=4.7Hz,2H,-CONHCH2-),3.46~3.90(m,10H,PEG-H),3.38(s,3H,-CH3)。
(2)E3的合成,用氢气将E2中的硝基还原为氨基,得E3;
所述步骤具体为:在一单口烧瓶中加入20ml甲醇,而后加入E2(113.0mg,0.32mmol),17.0mg Pd/C(10%)。通入H24h,温度为30℃,停止反应,硅藻土滤除Pd/C,得棕黄色油状液96.4mg,产率98%。
1H NMR(CDCl3,300MHz)δ6.78(s,2H,ArH),6.17(s,1H,ArH),4.41(t,J=4.5Hz,2H,-CONHCH2-),3.63~3.69(m,4H,-NH2),3.52~3.81(m,10H,PEG-H),3.36(s,3H,-CH3)。
(3)E4的合成,在缩合剂作用下,E3与酸反应得E4。
所述步骤具体为:在一单口烧瓶中加入20ml二氯甲烷,控制温度0℃,依次加入4-(三苯巯基)丁酸S3(280.3mg,0.81mmol),EDCl(192.0mg,0.97mmol),HOBt(131.2mg,0.97mmol),E3(80.5mg,0.27mmol),室温(30℃)下搅拌5h。停止反应,直接柱色谱分离,得棕黑色油状液65.0mg,产率40%。
1H NMR(CDCl3,300MHz)δ7.57(s,2H,ArH),7.23~7.42(m,30H,ArH),7.11(d,J=7.2Hz,1H,ArH),4.43(s,2H,-CONHCH2-),3.56~3.81(m,10H,PEG-H),3.37(s,3H,-CH3),2.20~2.30(m,8H,-NHCOCH2-和S-CH2-),1.65~1.78(m,4H,-CH2-)。
1.2当n=17时,亲水化合物E4的合成
其合成路径图如图1所示:
(1)E2的合成,在碱性条件下,取化合物E1与末端为氨基的PEG进行酰化反应,得化合物E2;
所述步骤具体为:在一单口烧瓶中加入100ml二氯甲烷,而后依次加入化合物末端为氨基的PEG750(12.0g,16.0mmol),三乙胺(3.34ml,24.0mmol),E1(5.53g,24.0mmol)控制0℃下搅拌10min。室温(25℃)下反应30min。停止反应以水洗涤,无水硫酸钠干燥,得红褐色液体15.37g。柱色谱分离,产率94.0%。
1H NMR(CDCl3,500MHz)δ9.23(t,J=2.0Hz,1H,ArH),9.19(d,J=2.0Hz,2H,ArH),4.60(t,J=4.7Hz,2H,-CONHCH2-),3.46~3.90(m,72H,PEG-H),3.38(s,3H,-CH3)。
(2)E3的合成,用氢气将E2中的硝基还原为氨基,得E3。
所述步骤具体为:在一单口烧瓶中加入50ml甲醇,而后加入E2(4.41g,4.70mmol),0.66gPd/C(10%)。通入H29h,温度为30℃,停止反应,硅藻土滤除Pd/C,得红褐色液4.10g,产率98.6%。
1H NMR(CDCl3,500MHz)δ6.76(d,J=1.9Hz,2H,ArH),6.19(t,J=1.9Hz,1H,ArH),4.40(t,J=4.9Hz,2H,-CONHCH2-),3.54~3.84(m,72H,PEG-H),3.38(s,3H,-CH3)。
(3)E4的合成,在缩合剂作用下,E3与酸反应得E4。
所述步骤具体为:在一单口烧瓶中加入30ml二氯甲烷,控制温度0℃,依次加入4-(三苯巯基)丁酸S3(0.83g,2.40mmol),EDCl(0.57g,2.88mmol),HOBt(0.39g,2.88mmol),E3(0.78g,0.8mmol),室温(30℃)下搅拌8h。停止反应,直接柱色谱分离,得红褐色油状液0.80g产率63%。
1H NMR(CDCl3,400MHz)δ7.96(s,2H,-NH-),7.77(s,2H,ArH),7.16~7.41(m,31H,ArH),4.43(t,J=4.8Hz,2H,-CONHCH2-),3.57~3.80(m,72H,PEG-H),3.37(s,3H,-CH3),2.26~2.30(m,8H,-NHCOCH2-和S-CH2-),1.74~1.78(m,4H,-CH2-)。
1.3当n=114时,亲水化合物E4的合成
其合成路径图如图1所示:
(1)E2的合成,在碱性条件下,取化合物E1与末端为氨基的PEG进行酰化反应,得化合物E2;
所述步骤具体为:在一100ml单口烧瓶中加入20mlDCM,而后依次加入末端为氨基的PEG5000(5.0g,1.0mmol)、NEt3(0.2ml,1.43mmol)和E1(0.92g,4.0mmol)。控制0℃下搅拌10min。然后室温下反应2h后停止反应。以水洗涤,无水硫酸钠干燥,得红褐色固体5.4g。柱色谱分离,产率91.0%。
1H NMR(CDCl3,500MHz)δ9.23(t,J=2.0Hz,1H,ArH),9.19(d,J=2.0Hz,2H,ArH),4.60(t,J=4.7Hz,2H,-CONHCH2-),3.46~3.90(m,452H,PEG-H),3.38(s,3H,-CH3)。
(2)E3的合成,用氢气将E2中的硝基还原为氨基,得E3。
所述步骤具体为:在一150ml单口烧瓶中加入E2(5.40g,1.04mmol),30ml甲醇,0.54g Pd/C(10%)。室温搅拌20min后通入H2,3h后停止反应,过滤除去Pd/C,得红褐色固体3.62g,产率90%。
1H NMR(CDCl3,400MHz)δ6.78(d,J=2.1Hz,2H,ArH),6.21(t,J=2.1Hz,1H,ArH),4.41(t,J=5.0Hz,2H,-CONHCH2-),3.79~3.83(m,4H,-NH2),3.53~3.72(m,452H,PEG-H),3.38(s,3H,-CH3);
(3)E4的合成,在缩合剂作用下,E3与酸反应得E4。
所述步骤具体为:在一50ml单口烧瓶中加入6mlDCM,4-(三苯巯基)丁酸S3(181.7mg,0.5mmol),EDCl(68.0mg,0.36mmol),HOBt(48.6mg,0.36mmol)控制温度在20℃以下。10min后加入E3(0.51g,0.1mmol),室温搅拌12h。停止反应,旋除DCM。柱色谱分离得红褐色固体产品0.36g,产率63%。
1H NMR(CDCl3,400MHz)δ7.99(s,2H,-NH-),7.91(s,2H,ArH),7.17~7.41(m,31H,ArH),4.52(t,J=4.8Hz,2H,-CONHCH2-),3.54~3.82(m,452H,PEG-H),3.38(s,3H,-CH3),2.26~2.30(m,8H,-NHCOCH2-和S-CH2-),1.74~1.78(m,4H,-CH2-)。
1.4当n=300时,亲水化合物E4的合成
其合成路径图如图1所示:
(1)E2的合成,在碱性条件下,取化合物E1与末端为氨基的PEG进行酰化反应,得化合物E2;
所述步骤具体为:在一100ml单口烧瓶中加入20ml二氯甲烷,而后依次加入末端为氨基的PEG(n=300)(13.2g,1.0mmol)、NEt3(0.2ml,1.43mmol)和E1(0.92g,4.0mmol)。控制0℃下搅拌10min。然后室温下反应2h后停止反应。以水洗涤,无水硫酸钠干燥,得红褐色固体5.4g。柱色谱分离,产率80.0%。
1H NMR(CDCl3,500MHz)δ9.21(t,J=2.0Hz,1H,ArH),9.17(d,J=2.0Hz,2H,ArH),4.61(t,J=4.7Hz,2H,-CONHCH2-),3.31~3.80(m,1195H,PEG-H),3.41(s,3H,-CH3)。
(2)E3的合成,用氢气将E2中的硝基还原为氨基,得E3。
所述步骤具体为:在一150ml单口烧瓶中加入E2(13.7g,1.04mmol),30ml甲醇,Pd/C(10%)。室温搅拌20min后通入H2,3h后停止反应,过滤除去Pd/C,得红褐色固体3.62g,产率78%。
1H NMR(CDCl3,400MHz)δ6.78(d,J=2.1Hz,2H,ArH),6.21(t,J=2.1Hz,1H,ArH),4.42(t,J=4.7Hz,2H,-CONHCH2-),3.79~3.83(m,4H,-NH2),3.50~3.79(m,1195H,PEG-H),3.38(s,3H,-CH3);
(3)E4的合成,在缩合剂作用下,E3与酸反应得E4。
所述步骤具体为:在一50ml单口烧瓶中加入6ml二氯甲烷,4-(三苯巯基)丁酸S3(109.0mg,0.3mmol),EDCl(68.0mg,0.36mmol),HOBt(48.6mg,0.36mmol)控制温度在20℃以下。10min后加入E3(0.51g,0.1mmol)),室温搅拌12h。停止反应,旋除DCM。柱色谱分离得红褐色固体产品,产率56%。
1HNMR(CDCl3,400MHz)δ7.91(s,2H,ArH),7.17~7.41(m,31H,ArH),4.53(t,J=4.7Hz,2H,-CONHCH2-),3.34~3.80(m,1195H,PEG-H),3.38(s,3H,-CH3),2.26~2.30(m,8H,-NHCOCH2-和S-CH2-),1.74~1.78(m,4H,-CH2-)。
实施例2、疏水化合物P1的合成
其中,R为烷基链。其合成路径图如图2所示:
2.1当R为碳十一烷基链时,疏水化合物P1的合成
(1)Z2的合成,在缩合剂作用下,通过Z1中的羧基和氨基反应得Z2。
所述步骤具体为:在一单口烧瓶中加入20mlDMF,控制温度0℃,而后依次加入化合物Z1(0.36g,2.0mmol),EDCl(0.95g,4.8mmol),HOBt(0.65g,4.8mmol),N2(2-(三苯巯基)乙胺)(1.91g,6.0mmol)。室温(25℃)下反应4h。停止反应以水洗涤,无水硫酸钠干燥。柱色谱分离,得淡黄色泡沫状固体3.7g,产率62%。
1H NMR(CDCl3,400MHz)δ7.17~7.42(m,31H,ArH),7.11(d,J=1.2Hz,2H,ArH),6.22(t,J=5.6Hz,2H,-NH-),3.90(s,2H,-NH2),3.23~3.28(dd,J1=6.0Hz,J2=12.4Hz,4H,-NCH2-),2.51(t,J=6.4Hz,4H,-SCH2-)。13C NMR(CDCl3,100MHz)δ167.25,147.67,145.03,136.08,129.75,128.45,127.06,116.54,114.68,67.17,38.63,32.07。
(2)P1的合成,在碱性条件下,氨基与酰氯反应得P1。
所述步骤具体为:在一单口烧瓶中加5ml二氯甲烷,而后依次加入化合物Z2(39.17mg,0.05mmol),三乙胺(0.02ml,0.15mmol),十二酰氯(0.024ml,0.1mmol)控制0℃下搅拌10min。室温(25℃)下反应30min。停止反应以水洗涤,无水硫酸钠干燥,得淡黄色泡沫状固体47.2mg,产率98%。
1H NMR(CDCl3,400MHz)δ8.75(s,1H,ArH),8.22(s,2H,ArH),7.78(s,1H,-NH-),7.15~7.40(m,30H,ArH),6.57(t,2H,J=5.6Hz,-NH-),3.22~3.27(dd,4H,J1=6.4Hz,J2=12.4Hz,-NCH2-),2.47(t,4H,J=6.4Hz,-SCH2-),2.34(t,2H,J=7.6Hz,-COCH2-),1.66(t,2H,J=7.2Hz,-CH2-),1.21~1.34(m,16H,-CH2CH2CH2-),0.87(t,3H,J=6.8Hz,-CH3)。13CNMR(CDCl3,100MHz)δ172.43,166.39,144.59,139.52,135.21,129.53,128.00,126.82,120.77,120.53,66.97,39.07,37.55,31.93,31.72,29.67,29.63,29.51,29.35,29.32,25.43,22.70,14.14.ESI-HRMS calc for【C62H66N3O3S2+H】+is 964.4573,found 964.4546.
实施例3、疏水化合物P2的合成
其中,R为 m为1~100中的任意一个整数。
疏水化合物P2的合成路径图如图3所示:
步骤一,制备化合物Z4
(1)Z3的合成,Z2与丁二酸酐在有机溶剂中反应得Z3。
所述步骤具体为:以CHCl3做溶剂,Z2和丁二酸酐在常温下反应,TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色泡沫状固体产品。其中所述的Z2(mol)和丁二酸酐(mol)之比为1∶1.5~2。
1HNMR(d6-DMSO,400MHz)δ10.38(s,1H,-COOH),8.60(s,2H,-NH-),7.82(s,1H,-NH-),7.17~7.40(m,32H,ArH)3.16~3.19(m,4H,-NCH2-),2.47(t,J=1.6Hz,4H,-SCH2-),2.31(t,J=7.2Hz,4H,-COCH2-)。13C NMR(CDCl3,100MHz)δ180.47,171.70,166.26,144.91,139.92,135.54,129.54,128.51,127.21,121.02,120.61,66.44,38.77,31.69,25.57,21.23。ESI-HRMS calc for【C54H49N3O5S2+Na】+is 906.3011,found 906.2995.
(2)Z4的合成,在缩合剂作用下,Z3与丙炔胺反应得Z4。
所述步骤具体为:以DCM做溶剂,控制温度0℃,先用EDCl、HOBt把Z3活化完全,以活化后的物质和丙炔胺在常温(30℃)反应。TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色泡沫状固体产品。其中所述的EDCl(mol),HOBt(mol),Z3(mol)和丙炔胺(mol)之比为1.2∶1.2∶1∶4。
1H NMR(CDCl3,400MHz)δ9.29(s,1H,ArH),7.58(s,1H,ArH),7.12~7.46(m,30H,ArH),6.86(t,2H,J=5.2Hz,-NH-),6.03(s,1H,-NH-),3.85(t,2H,J1=2.4Hz,-CH2-),3.24(dd,4H,J1=6.4Hz,J2=12.0Hz,-NCH2-),2.77(t,2H,J=6.0Hz,-CH2-),2.56(t,2H,J=5.6Hz,-CH2-),2.50(t,4H,J=6.8Hz,-SCH2-),2.05(s,1H,炔氢)。13C NMR(CDCl3,100MHz)δ172.57,171.49,166.72,144.72,138.35.135.40,129.63,128.01,126.83,120.96,120.49,79.16,71.71,66.82,39.11,31.93,31.41,30.27,29.71。ESI-HRMS calc for【C57H52N4O4S2+Na】+is 943.3328,,found 943.3334.
步骤二,取聚乳酸,其中m为1~100的任一整数,制备得到A3
当聚乳酸中m=2时,A3的制备。
(1)A2的合成,A1与氯乙酰氯反应得A2。
所述步骤具体为:以DCM做溶剂,A1在NEt3的作用下,和氯乙酰氯在0℃下反应,TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得淡黄色油状液体产品。其中所述的A1(mol),NEt3(mol)和氯乙酰氯(mol)之比为1∶6∶10。
1H NMR(CDCl3,400MHz)δ7.31~7.39(m,5H,ArH),5.13~5.25(m,4H,-CH2-,-CH-),4.13(s,2H,-CH2-Cl),1.53~1.55(m,6H,-CH3)。IR(KBr)3671.04,2961.70,1750.11,1453.23,1410.90,1260.25,1092.16,1020.60,866.98,798.74,697.62。
(2)A3的合成,A2与叠氮钠反应得A3。
所述步骤具体为:以DMSO做溶剂,A2和NaN3在常温下反应,TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得淡黄色油状液体产品。其中所述的A2(mol)和NaN3(mol)之比为1∶5~10。
IR(KBr)3448.21,2929.30,2103.57,1643.16,1381.62,1177.24,1088.10,693.73。
当聚乳酸中m=4时,A3的制备。
(1)A2的合成,A1与氯乙酰氯反应得A2。
所述步骤具体为:在一单口烧瓶中加入5ml DCM,而后依次加入化合物A1(79.23mg,0.2mmol),三乙胺0.073ml,冰浴条件下滴加氯乙酰氯0.079ml,10min后撤去冰浴,室温(25℃)下过夜,停止反应以水洗涤,无水硫酸钠干燥,柱分离得35.0mg无色油状液,产率64%。
1H NMR(CDCl3,400MHz)δ7.31~7.38(m,5H,ArH),5.12~5.30(m,6H,-CH2-,-CH-),4.14(s,2H,-CH2-Cl),1.50~1.61(m,12H,-CH3)。IR(KBr)2994.33,2944.70,1746.73,1498.97,1453.68,1269.40,1187.60,1091.02,1044.73,962.11,789.02,752.21,699.82.
(2)A3的合成,A2与叠氮钠反应得A3。
所述步骤具体为:单口瓶中加入5mlDMSO,A2(30.0mg,0.064mmol)室温搅拌,30min后加入NaN3(20.66mg,0.32mmol),反应2d,停止反应以水洗涤,DCM萃取,无水硫酸钠干燥,得淡黄色油状液15.0mg,产率50.0%。
IR(KBr)2993.55,2943.45,2106.19,1745.90,1498.81,1452.25,1270.24,1181.09,1128.37,1087.33,1042.72,739.25,697.55。
当聚乳酸中m=100时,A3的制备。
(1)A2的合成,A1与氯乙酰氯反应得A2。
所述步骤具体为:以DCM做溶剂,A1在NEt3的作用下,和氯乙酰氯在0℃下反应,TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得淡黄色油状液体产品。其中所述的A1(mol),NEt3(mol)和氯乙酰氯(mol)之比为1∶6∶10。
1H NMR(CDCl3,400MHz)δ7.31~7.41(m,5H,ArH),5.10~5.25(m,102H,-CH2-,-CH-),4.13(s,2H,-CH2-Cl),1.50~1.62(m,300H,-CH3)。IR(KBr)3671.04,2961.70,1750.11,1453.23,1410.90,1260.25,1092.16,1020.60,866.98,798.74,697.62。
(2)A3的合成,A2与叠氮钠反应得A3。
所述步骤具体为:以DMSO做溶剂,A2和NaN3在常温下反应,TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得淡黄色油状液体产品。其中所述的A2(mol)和NaN3(mol)之比为1∶5~10。
IR(KBr)3448.21,2929.30,2103.57,1643.16,1381.62,1177.24,1088.10,693.73。
步骤三,将A3与Z4反应制备得到P2。
当A3中m=2时,
所述步骤具体为:在两口瓶中,将等当量的Z4和A3溶于适量THF,使得两化合物的浓度为10mmol/ml。体系抽充氮气三次,加氮气球,使反应在氮气保护下进行。将0.6倍当量的无水CuSO4固体和2倍当量的抗坏血酸钠(VcNa)混合,抽真空,加入去离子水震荡得黄色悬浊液,再注入反应体系中,室温搅拌36h。反应结束后加入适量DCM萃取体系,水洗后无水硫酸钠干燥,柱分离,产率52%。
1H NMR(CDCl3,400MHz)δ9.44(s,1H,-NH-),7.72(s,2H,ArH),7.58(s,1H,=CH-,),7.53(s,1H,ArH),7.24~7.43(m,30H,ArH),7.17~7.21(m,5H,ArH),6.92(t,2H,J=5.2Hz,-NH-),6.77(t,1H,J=5.6Hz,-NH-),5.13~5.21(m,6H,-CH2-and-CH-),4.41(d,J=5.2Hz,2H,-CH2-),3.22~3.26(dd,4H,J1=6.0Hz,J2=12.0Hz,-NCH2-),2.66(t,2H,J=6.8Hz),2.48~2.54(m,6H,-SCH2-and-CH2-),1.45~1.57(m,6H,-CH3-)。13C NMR(CDCl3,100MHz)δ172.61,171.41,169.85,169.25,166.72,166.16,144.70,138.75,135.41,135.03,129.59,128.67,128.59,128.25,127.99,126.79,123.86,120.79,120.56,70.00,69.48,50.43,39.09,34.96,31.80,30.58,29.71,16.76,16.59.ESI-HRMScalc for【C72H69N7O10S2+H】+is 1256.4678,found 1256.4626。
当A3中m=4时,
所述步骤具体为:在两口瓶中,将等当量的Z4和A3溶于适量THF,使得两化合物的浓度为10mmol/ml。体系抽充氮气三次,加氮气球,使反应在氮气保护下进行。将0.6倍当量的无水CuSO4固体和3倍当量的抗坏血酸钠(VcNa)混合,抽真空,加入去离子水震荡得黄色悬浊液,再注入反应体系中,室温搅拌36h。反应结束后加入适量DCM萃取体系,水洗后无水硫酸钠干燥,柱分离,产率55%。
1HNMR(CDCl3,400MHz)δ9.35(s,1H,ArH),7.76(s,1H,ArH),7.60(s,1H,-NH-),7.25~7.43(m,30H,ArH),7.18~7.21(m,6H,ArH和烯氢),6.89(t,2H,J=5.2Hz,-NH-),6.77(s,1H,-NH-),5.12~5.24(m,8H,-CH2-和-CH-),4.44(d,2H,-CH2-),3.23~3.27(dd,4H,J1=6.4Hz,J2=12.0Hz,-NCH2-),2.65(t,2H,J=8.4Hz),2.49~2.67(m,6H,-SCH2-和-CH2-),1.51~1.59(m,12H,-CH3-)。13C NMR(CDCl3,100MHz)δ169.94,169.53,169.37,169.30,166.61,166.08,144.69,138.72,135.38,135.06,129.58,128.64,128.54,128.24,127.99,126.79,120.64,120.62,69.92,69.39,69.32,67.25,66.86,50.43,39.08,35.02,31.79,30.74,29.70,16.73,16.70,16.65,14.20.ESI-HRMS calc for【C78H78N7O14S2+H】+is 1400.5062,found 1400.5048.
当A3中m=100时,
所述步骤具体为:在两口瓶中,将等当量的Z4和A3溶于适量THF,使得两化合物的浓度为10mmol/ml。体系抽充氮气三次,加氮气球,使反应在氮气保护下进行。将0.6倍当量的无水CuSO4固体和2倍当量的抗坏血酸钠(VcNa)混合,抽真空,加入去离子水震荡得黄色悬浊液,再注入反应体系中,室温搅拌36h。反应结束后加入适量DCM萃取体系,水洗后无水硫酸钠干燥,柱分离,产率43%。
δ9.44(s,1H,-NH-),7.72(s,2H,ArH),7.58(s,1H,=CH-,),7.53(s,1H,ArH),7.24~7.43(m,30H,ArH),7.17~7.23(m,5H,ArH),6.92(t,2H,J=5.2Hz,-NH-),6.77(t,1H,J=5.6Hz,-NH-),5.13~5.22(m,103H,-CH2-and-CH-),4.41(d,J=5.2Hz,2H,-CH2-),3.22~3.28(dd,4H,J1=6.0Hz,J2=12.0Hz,-NCH2-),2.66(t,2H,J=6.8Hz),2.39~2.56(m,6H,-SCH2-and-CH2-),1.45~1.57(m,300H,-CH3-)。
实施例4、疏水化合物P3
其中, 时,k为1~100中的任一整数。
其合成路径图如图4所示:
当k=1时,
(1)B2的合成,B1与丁二酸酐反应得B2。
所述步骤具体为:以DMF做溶剂,B1在DMAP的作用下,和丁二酸酐在室温反应。TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色粘稠的液体产品。其中所述的B1(mol),DMAP(mol)和丁二酸酐(mol)之比为1∶5∶1~2。该反应产物未经表征,直接用于下一步反应。
(2)P3的合成,在缩合剂作用下,B2与Z2反应得P3。
所述步骤具体为:在一单口烧瓶中加入DMF,控制温度0℃,而后依次加入化合物B2(0.3mmol)、EDCl(68.7mg,0.36mmol)和HOBt(48.6mg,0.36mmol),活化2h,然后加入Z2(78.3mg,0.1mmol)。室温(25℃)下反应10h。停止反应以水洗涤,无水硫酸钠干燥。柱色谱分离,得无色液体,产率75%。
1HNMR(CDCl3,400MHz)δ8.06(d,J=14.8Hz,2H,ArH),7.81(s,1H,ArH),7.19~7.44(m,30H,ArH),6.54(t,J=1.2Hz,2H,-NH-),5.13~5.25(m,2H,-CH-),4.18(s,4H,-CH2-),3.27~3.32(dd,4H,J1=6.4Hz,J2=12.0Hz,-NCH2-),2.72~2.82(m,8H,-CH2-),2.52(t,J=6.4Hz,4H,-SCH2-),1.47~1.61(m,6H,-CH3)。
当k=21时,
(1)B2的合成,B1与丁二酸酐反应得B2。
所述步骤具体为:以DMF做溶剂,B1在DMAP的作用下,和丁二酸酐在室温反应。TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色粘稠的液体产品。其中所述的B1(mol),DMAP(mol)和丁二酸酐(mol)之比为1∶5∶1~2。该反应产物未经表征,直接用于下一步反应。
(2)P3的合成,在缩合剂作用下,B2与Z2反应得P3。
所述步骤具体为:在一单口烧瓶中加入DMF,控制温度0℃,而后依次加入化合物B2(960mg,0.3mmol)、EDCl(68.7mg,0.36mmol)和HOBt(48.6mg,0.36mmol),活化2h,然后加入Z2(78.3mg,0.1mmol)。室温(25℃)下反应12h。停止反应以水洗涤,无水硫酸钠干燥。柱色谱分离,得白色泡沫状固体260mg。产率65%。
1H NMR(CDCl3,400MHz)δ8.06(d,J=14.8Hz,2H,ArH),7.81(s,1H,ArH),7.19~7.44(m,30H,ArH),6.54(t,J=1.2Hz,2H,-NH-),5.13~5.25(m,42H,-CH-),4.18(s,4H,-CH2-),3.27~3.32(dd,4H,J1=6.4Hz,J2=12.0Hz,-NCH2-),2.72~2.82(m,8H,-CH2-),2.52(t,J=6.4Hz,4H,-SCH2-),1.47~1.61(m,126H,-CH3)。
当k=100时,
(1)B2的合成,B1与丁二酸酐反应得B2。
所述步骤具体为:以DMF做溶剂,B1在DMAP的作用下,和丁二酸酐在室温反应。TLC跟踪至反应完全。反应结束后,加入适量水,用DCM萃取,有机相分别用水、饱和食盐水洗,无水硫酸钠干燥,减压旋去溶剂,柱层析得白色粘稠的液体产品。其中所述的B1(mol),DMAP(mol)和丁二酸酐(mol)之比为1∶5∶1~2。该反应产物未经表征,直接用于下一步反应。
(2)P3的合成,在缩合剂作用下,B2与Z2反应得P3。
所述步骤具体为:在一单口烧瓶中加入DMF,控制温度0℃,而后依次加入化合物B2(960mg,0.3mmol)、EDCl(68.7mg,0.36mmol)和HOBt(48.6mg,0.36mmol),活化2h,然后加入Z2(78.3mg,0.1mmol)。室温(25℃)下反应18h。停止反应以水洗涤,无水硫酸钠干燥。柱色谱分离,得白色固体,产率54%。
1H NMR(CDCl3,400MHz)δ8.06(d,J=14.8Hz,2H,ArH),7.81(s,1H,ArH),7.19~7.44(m,30H,ArH),6.54(t,J=1.2Hz,2H,-NH-),5.13~5.25(m,200H,-CH-),4.18(s,4H,-CH2-),3.27~3.32(m,4H),2.72~2.82(m,8H,-CH2-),2.52(t,J=6.4Hz,4H,-SCH2-),1.45~1.66(m,600H,-CH3)。
实施例5、两亲性嵌段共聚物M的合成
5.1当PEG的n=17,R=碳十一烷基链时,M1的合成。
M1的合成路径图如图5所示,分别取亲水化合物、疏水化合物和氧化剂,在卤代烃溶剂中进行混合,即得所述化合物M1。
所述步骤具体为:分别取30ml E4和P1的二氯甲烷混合溶液(2.0mM)置于一250ml单口瓶中,其中E4和P1的摩尔比可以为1∶1~10,混合均匀后旋除二氯甲烷,残留物用10mL~200mL I2的二氯甲烷溶液(6.0mM)溶解,常温搅拌1h后将反应液冷却到0℃,加入Na2S2O3(3.0mM)直至I2的颜色消失。有机层用饱和NaCl(aq)洗涤,无水硫酸钠干燥,柱色谱分离,得淡黄色固体M168mg,产率64%。
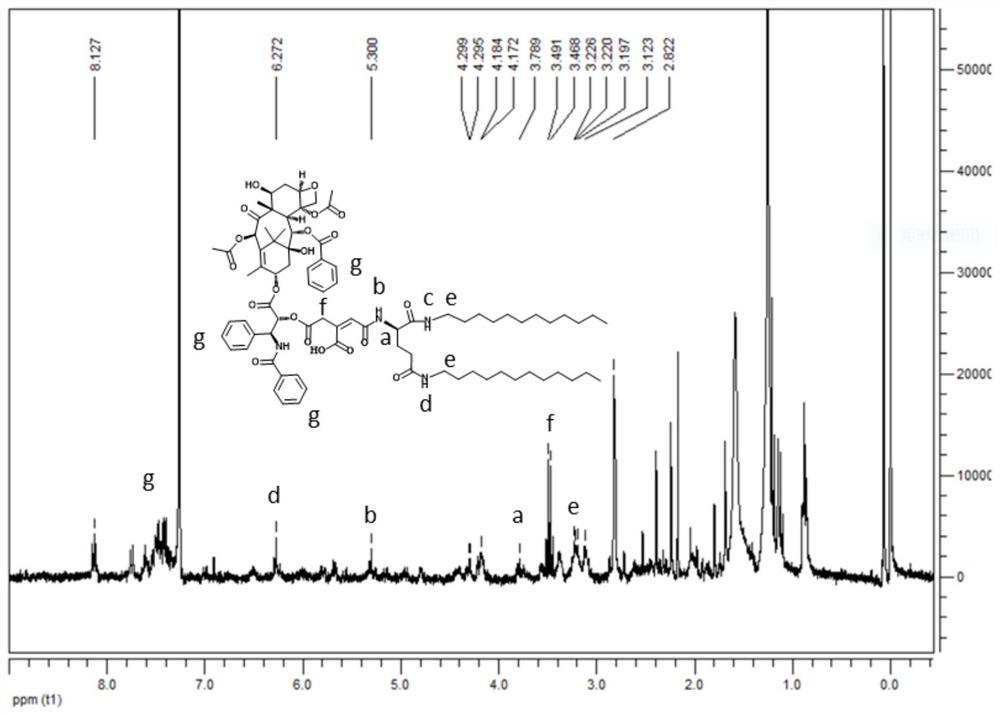
1HNMR(CDCl3,500MHz)δ9.23(s,2H,ArH),8.97(s,1H,ArH),8.08(s,5H,-NH-),7.86(s,2H,ArH),7.47(s,1H,ArH),4.34(t,J=4.4Hz,2H,-CO2CH2-),3.52-3.75(m,72H,PEG-H),3.33(s,3H,PEG-CH3),2.81~2.87(m,8H,-CONHCH2-和-SCH2-),2.59(t,J=1.2Hz,4H,-NCH2-),2.29(t,J=7.6Hz,2H,-COCH2-),2.05(t,J=6.8Hz 4H,-CH2-),1.66(t,J=7.2Hz 2H,-CH2-),1.21~1.25(m,16H,-CH2CH2CH2-),0.85(t,J=6.8Hz 3H,-CH3)。
从图8的E4,P1和M1的红外谱图可以看出M1的IR谱图是E4和P1的IR谱图的综合。在M1的IR谱图中,1081cm-1处是C-O-C的伸缩振动峰,1346cm-1处是甲基的特征峰,1694cm-1处为羰基的对称伸缩振动吸收峰,2919cm-1,2852cm-1处是烷基C-H伸缩振动产生的吸收峰,3290cm-1,1604cm-1,1446cm-1处这些峰说明有苯环存在,其中3290cm-1处的吸收峰为苯环上C-H伸缩振动产生的吸收峰,后三个峰是苯环碳骨架伸缩振动的特征吸收峰,1259cm-1~947cm-1处的峰为Ar-H面内弯曲振动产生的,770cm-1和684cm-1为以取代芳环的Ar-H面外弯曲振动产生的,720cm-1处是4个以上CH2成直链时其C-H面外弯曲振动产生的吸收峰。说明E4和P1在I2的DCM溶液中生成了新型两亲性嵌段共聚物M1。
表1M1的分子量和分子量分布范围
从图9和表1可以看出,嵌段共聚物的最大保留时间相对亲水片段和疏水片段缩短,说明形成了两亲性嵌段共聚物。两亲性嵌段共聚物形成之后,共聚物的分子量分布也有变化,在共聚物M1中D由1.03和1.10变为1.20.所合成的聚合物的相对分子质量分布比较窄。
5.2当PEG的n=17,
其中m=2时,M2的合成:
M2的合成路径图如图5所示,分别取亲水化合物、疏水化合物和氧化剂,在卤代烃溶剂中进行混合,即得所述化合物M2。
所述步骤具体为:分别取30ml E4和P2的二氯甲烷溶液(2.0mM)置于一250ml单口瓶中,混合均匀旋除DCM,残留物用120ml I2的二氯甲烷溶液(6.0mM)溶解,常温搅拌10h后将反应液冷却到0℃,加入Na2S2O3(3.0mM)直至I2的颜色消失。有机层用饱和NaCl(aq)洗涤,无水硫酸钠干燥,柱色谱分离,得淡黄色固体M2,产率62%。
1HNMR(CDCl3,400MHz)δ9.76(s,1H,ArH),9.57(s,1H,ArH),9.15(s,1H,ArH),8.34(s,1H,ArH),8.00(s,1H,ArH),7.81(s,1H,ArH),7.29~7.36(m,6H,ArH andCH=),5.10~5.22(m,6H,-CH-and-CH2-),4.49(s,2H,-CH2-),4.34(m,2H,-CO2CH2-),3.54~3.80(m,72H,PEG-H),3.38(s,3H,PEG-CH3),2.53~3.01(m,24H,-CH2-),2.05~2.13(m,4H,-CH2-),1.51~1.55(m,6H,-CH3)。
红外谱图如图10的E4,P2和M2的谱图,可以看出M2的IR谱图是E4和P2的IR谱图的叠加。在M2的IR谱图中,1081cm-1处是C-O-C的伸缩振动峰,1345cm-1处是甲基的特征峰,1750cm-1为羰基的对称伸缩振动吸收峰,2916cm-1,2887cm-1是烷基C-H伸缩振动产生的吸收峰,3285cm-1,1610cm-1,1447cm-1处这些峰说明有苯环存在,其中3285cm-1处的吸收峰为苯环上C-H伸缩振动产生的吸收峰,后三个峰是苯环碳骨架伸缩振动的特征吸收峰,1301cm-1~950cm-1处的峰为Ar-H面内弯曲振动产生的,742cm-1为一取代芳环的Ar-H面外弯曲振动产生的,说明E4和P2在I2的DCM溶液中生成了两亲性嵌段共聚物M2。
表2M2的分子量和分子量分布范围
从图11和表2可以看出,嵌段共聚物的最大保留时间相对亲水片段和疏水片段缩短,说明形成了两亲性嵌段共聚物。两亲性嵌段共聚物形成之后,共聚物的分子量分布也有变化,在共聚物M2中D由1.03和1.10变为1.13.所合成的聚合物的相对分子质量分布比较窄。
5.3当PEG的n=17,
其中m=4时,M3的合成:
M3的合成路径图如图5所示,分别取亲水化合物、疏水化合物和氧化剂,在卤代烃溶剂中进行混合,即得所述化合物M3。
所述步骤具体为:分别取30ml E4和P2的二氯甲烷溶液(2.0mM)置于一250ml单口瓶中,混合均匀旋除DCM,残留物用120ml I2的二氯甲烷溶液(6.0mM)溶解,常温搅拌10h后将反应液冷却到0℃,加入Na2S2O3(3.0mM)直至I2的颜色消失。有机层用饱和NaCl(aq)洗涤,无水硫酸钠干燥,柱色谱分离,得淡黄色固体M3,产率60%。
1HNMR(CDCl3,400MHz)δ9.77(s,1H,ArH),9.52(s,1H,ArH),9.01(s,1H,ArH),8.20(s,1H,ArH),7.94(s,2H,ArH),7.32~7.34(m,6H,ArH and CH=),5.09~5.24(m,6H,-CH-and-CH2-),4.49~4.50(m,2H,-CH2-),4.38~4.40(m,2H,-CO2CH2-),3.55~3.62(m,72H,PEG-H),3.37(s,3H,PEG-CH3),2.50~3.09(m,24H,-CH2-),2.11(m,4H,-CH2-),1.50~1.56(m,12H,-CH3)。
图12为E4,P2和M3的红外谱图可以看出M3的IR谱图是E4和P2的IR谱图的综合。在M3的IR谱图中,1097cm-1处是C-O-C的伸缩振动峰,1350cm-1处是甲基的特征峰,1740cm-1为羰基的对称伸缩振动吸收峰,2921cm-1,2889cm-1是烷基C-H伸缩振动产生的吸收峰,3306cm-1,1605cm-1,1450cm-1处这些峰说明有苯环存在,其中3306cm-1处的吸收峰为苯环上C-H伸缩振动产生的吸收峰,后三个峰时苯环碳骨架伸缩振动的特征吸收峰,1181cm-1~952cm-1处的峰为Ar-H面内弯曲振动产生的,952cm-1和733cm-1为一取代芳环的Ar-H面外弯曲振动产生的,说明E4和P2在I2的DCM溶液中生成了两亲性嵌段共聚物M3。
表3M3的分子量和分子量分布范围
从图13和表3可以看出,嵌段共聚物的最大保留时间相对亲水片段和疏水片段缩短,说明形成了两亲性嵌段共聚物。两亲性嵌段共聚物形成之后,共聚物的分子量分布也有变化,在共聚物M3中D由1.03和1.10变为1.29.所合成的聚合物的相对分子质量分布比较窄。
5.4当PEG的n=114,
其中k=21时,M4的合成:
M4的合成路径图如图5所示,分别取亲水化合物、疏水化合物和氧化剂,在卤代烃溶剂中进行混合,即得所述化合物M4。
所述步骤具体为:分别取30ml E4和P3的二氯甲烷溶液(2.0mM)置于一250ml单口瓶中,混合均匀旋除DCM,残留物用120ml I2的二氯甲烷溶液(6.0mM)溶解,常温搅拌10h后将反应液冷却到0℃,加入Na2S2O3(3.0mM)直至I2的颜色消失。有机层用饱和NaCl(aq)洗涤,无水硫酸钠干燥,柱色谱分离,得淡黄色固体M4,产率62%。1H NMR(CDCl3,400MHz)δ9.15(s,2H,ArH),8.96(s,1H,ArH),8.17(s,2H,ArH),7.17(s,1H,ArH),5.13~5.15(m,29H,PLA-CH-),4.1
通过分子胶连接的两亲性嵌段共聚物及其合成方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




![吡咯[2,1-a]异喹啉生物碱及其制备方法和应用](https://www.zhichawang.com/images/CN107522697A/CN107522697A.jpg)











动态评分
0.0