








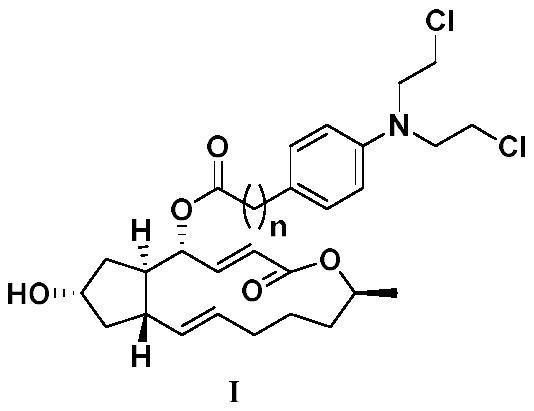
专利摘要
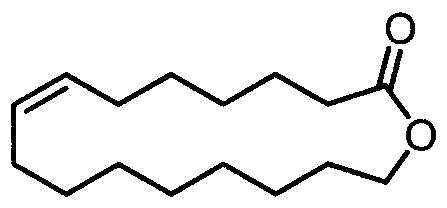
本发明公开了一种11‑十一内酯类化合物和己内酯类化合物的制备方法,该方法以环己酮类螺环过氧化物为原料,采用质子酸为催化剂,氟醇为溶剂,反应温度为25℃~60℃。本发明具有收率高、成本低廉、操作方便、反应条件温和等优点,便于工业化应用。
权利要求
1.一种11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,包括:
在质子酸的催化下,环己酮类螺环过氧化物在氟醇溶剂中进行重排反应,得到11-十一内酯类化合物和己内酯类化合物;
所述的11-十一内酯类化合物的结构如下式:
所述的环己酮类螺环过氧化物的结构如式(1)所示:
式(1)中,R
所述的氟醇溶剂的结构如式(3)所示:
式(3)中,X、Y独立地选自F或H;
Rf为碳原子数为1~7的全氟烷基。
2.根据权利要求1所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,所述的R
3.根据权利要求1所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,式(3)中,X、Y都为H。
4.根据权利要求1所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,所述的质子酸为无机强酸、有机强酸和超强酸中的一种或者多种;
以环己酮类螺环过氧化物计,所述的质子酸的用量为1~5mol%。
5.根据权利要求4所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,所述的无机强酸为硫酸、硝酸、高氯酸、盐酸、氢溴酸、氢碘酸、高溴酸、氯酸、溴酸、氟硅酸、氯铅酸、偏磷酸、锇酸、高锰酸、硒酸、高铁酸、氟硼酸、氰酸中的一种或者多种;
所述的有机强酸为2,4,6-三硝基苯酚、2,4,6-三硝基苯甲酸、三氟乙酸、三氯乙酸、对甲基苯磺酸、甲磺酸、苯磺酸、苯胂酸、KMD酸、苯膦酸中的一种或多种;
所述的超强酸为氟锑酸、氟锑磺酸、全氟磺酸树脂、氯氟铝酸、碳硼烷酸、特里布尔挥发酸、ZXQ酸、固体超强酸中的一种或多种。
6.根据权利要求5所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,所述的无机强酸为盐酸。
7.根据权利要求5所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,所述的有机强酸为对甲基苯磺酸。
8.根据权利要求1所述的11-十一内酯类化合物和己内酯类化合物的制备方法,所述的重排反应的温度为25℃~60℃;
所述的环己酮类螺环过氧化物与氟醇溶剂的摩尔体积比为0.01mol/L~0.5mol/L;
所述的重排反应的反应时间为10min~60min。
9.根据权利要求1所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,还包括环己酮类螺环过氧化物的制备步骤:
所述的环己酮类螺环过氧化物由环己酮类化合物和双氧水制备得到。
10.根据权利要求1所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,所述的11-十一内酯类化合物和己内酯类化合物通过减压精馏进行分离。
11.一种如权利要求1~10任一项所述的11-十一内酯类化合物和己内酯类化合物的制备方法,其特征在于,所述的氟醇溶剂替换为1,2,2-三氟乙醇、2,2,2-三氟乙醇、1,1,2,2,2-五氟乙醇、2,2,3,3-四氟丙醇、2,2,3,3,3-五氟丙醇、1,1,2,2-四氟丁醇、1,1,2,2-四氟戊醇、1,1,2,2-四氟己醇、1,1,2,2,3,3-六氟己醇、1,1,2,2-四氟庚醇、1,1,2,2,3,3-六氟庚醇、2,2,2,3,3-五氟辛醇中的一种或者多种。
说明书
技术领域
本发明属于有机合成领域,具体涉及一种由环己酮类螺环过氧化物制备11-十一内酯类化合物和己内酯类化合物的方法。
背景技术
大环内酯是一种重要的化工中间体,经常被用作麝香香料。传统的天然麝香一般是从动物身上获得,由于来源有限而且经常要以动物的死亡为前提,其价格极为昂贵,且远远不能满足人们的需求。在人工合成的麝香香料中,硝基麝香为人类首先合成得到的具有麝香气味的化学品,其价格低廉,市场占比最大,但由于其对皮肤有光敏作用,存在致癌风险,逐渐被淘汰。其后多环麝香由于其廉价、稳定、方便使用的优点,逐渐开始取代硝基麝香并成为工业标准。然而,相对于该两类麝香,大环麝香(大环内酯)是结构上最接近天然麝香的产品,其香气更为细腻持久,具有很强的天然感,常用于调配麝香系列的高级香精,具有良好的定香作用,也用于调配红甘草、欧黑莓、番石榴等食用香精。但是大环内酯的制备工艺一般都更为复杂,原料有限、合成路线较长、收率低导致成本更为昂贵,在商品化麝香中占份额较小。目前,每年工业合成的大环内酯麝香大约为100吨,因此开发步骤少、收率高的大环麝香路线具有重大的现实意义。
除了在香精香料领域,大环内酯在医药领域也有广泛的应用,例如抗生素红霉素等。近些年来,人工合成大环内酯领域不断取得进步,工业上的制备方法一般分为五种:(1)从环十二酮出发的环扩张反应(如专利JP Patent 53015344),该方法为目前工业上最经济的十五内酯制备方法。原料环十二酮一般通过丁二烯三聚等多步反应获得(Scheme 1),年产量约为每年10万吨,此外环扩张的工艺中一般需要用到强氧化剂如臭氧、单线态氧,或强还原剂如硼氢化钠、氢化铝锂等,对设备要求较高,并存在不可忽视的安全问题。
Scheme 1.通过丁二烯合成环十二酮
(2)酮醇缩合反应(如文献Helv.Chim.Acta1947,30,1815),该方法为第一个工业化制备麝香酮和环十五烷酮的方法,但该方法使用金属钠作为还原剂,并不是一个安全绿色的反应;
(3)聚合-解聚反应(如专利US Patent 2020298),杜邦公司的Carothers于1930年代首先提出了该方法,此后多种适用于该方法的催化剂被发明出来,该方法最大的缺点为原料1,5-羟基十酸不易得到,工业上一般是通过单酯化己二酸盐与11-羟基十一烷酸电化学Kolbe偶联获得;
(4)酯基转移反应(如专利US Patent 2417151),该方法由Collaud于1941年提出,使用与聚合-解聚工艺同样的15-羟基十酸为底物。
(5)烷基过氧化物热分解反应,又称为Story合成(Scheme 2)(如专利US Patent3776926),该方法原料廉价且来源广泛,同时反应步骤较少,不过由于中间体环己酮类螺环过氧化物在温度较高时存在爆炸的危险,并且反应选择性不够高,限制了其应用,若能在操作安全性和反应选择性上进行改进,对其工业上的应用将具有重要的意义;
Scheme 2.通过Story法合成大环化合物
另外值得一提的是,2001年Albrecht Berkessel(Angew.Chem.Int.Ed.2002,41,4481.)提出了在六氟异丙醇(HFIP)中合成环己酮螺环过氧化物的方法(Scheme 3),该方法环己酮类螺环过氧化物的收率较Story方法有很大的提高,该中间体收率的提高使该Story方法离工业应用更进了一步。
Scheme 3.在HFIP中合成环己酮类螺环过氧化物
发明内容
本发明提供了一种11-十一内酯类化合物和己内酯类化合物的制备方法,该方法反应条件温和,明显降低了Story方法中间体爆炸的风险;同时提高了产物的选择性和收率。
本发明的技术方案为:
一种11-十一内酯类化合物和己内酯类化合物的制备方法,包括:
在质子酸的催化下,环己酮类螺环过氧化物在氟醇溶剂中进行重排反应,得到11-十一内酯类化合物和己内酯类化合物;
所述的环己酮类螺环过氧化物的结构如式(1)所示:
式(1)中,Rn为H或C1~C5烷基。
本发明的反应方程式如下:
本发明以环己酮类螺环过氧化物作为原料,以质子酸和氟醇溶剂作为反应介质,同现有技术相比,一方面降低了反应温度(由150℃至少降低至60℃),极大地减少了过氧化物爆炸的风险,另一方面,明显提高了11-十一内酯的收率;此外,该方法产生的另一产物己内酯类化合物为重要的聚合物单体,具有较大的应用意义。
其中,所述的Rn为H、甲基、乙基、丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基。
本发明中,氟醇溶剂的种类会对重排反应产生很大的影响,该氟醇溶剂需要具有一定给氢键能力,不过其给氢键能力弱于六氟异丙醇。本发明的氟醇溶剂具有如式(3)所示的通式:
式(3)中,X、Y独立地选自F或H;
Rf为被至少一个氟原子取代的C1~C7烷基。
本发明的氟醇溶剂具体可以选自以下物质中的一种或者多种:1,2,2-三氟乙醇、2,2,2-三氟乙醇、1,1,2,2,2-五氟乙醇、2,2,3,3-四氟丙醇、2,2,3,3,3-五氟丙醇、1,1,2,2-四氟丁醇、1,1,2,2-四氟戊醇、1,1,2,2-四氟己醇、1,1,2,2,3,3-六氟己醇、1,1,2,2-四氟庚醇、1,1,2,2,3,3-六氟庚醇、2,2,2,3,3-五氟辛醇。
作为优选,氟醇溶剂可以采用下述式(2)表示的直链氟醇溶剂,Rf为被至少一个氟原子取代的C1~C7烷基;进一步优选的,Rf为1~7的全氟烷基。所述的氟醇溶剂最优选为2,2,2-三氟乙醇。
(式中,Rf表示碳原子数为1~7的全氟烷基)。
本发明中,所用的催化剂为质子酸,可以是无机强酸,例如硫酸、硝酸、高氯酸、盐酸、氢溴酸、氢碘酸、高溴酸、氯酸、溴酸、氟硅酸、氯铅酸、偏磷酸、锇酸、高锰酸、硒酸、高铁酸、氟硼酸、氰酸、硫氰偏高碘酸等中的一种或者多种,其中优选的为盐酸。也可以是有机强酸,例如2,4,6-三硝基苯酚、2,4,6-三硝基苯甲酸、三氟乙酸、三氯乙酸、对甲基苯磺酸、甲磺酸、苯磺酸、苯胂酸、KMD酸、苯膦酸等中的一种或者多种,其中最为优选的为对甲基苯磺酸。还可以是超强酸,例如氟锑酸、氟锑磺酸、全氟磺酸树脂、氯氟铝酸、碳硼烷酸、特里布尔挥发酸、ZXQ酸、固体超强酸等中的一种或者多种。
本发明中,存在一定量的水不会对反应产生不利的影响,因此,可以直接采用工业上容易获得的一水对甲基苯磺酸和盐酸水溶液等直接进行投料。
本发明中,所述的催化剂浓度优选为1~5mol%,反应温度为25℃~60℃,优选的温度为室温25℃,此时,不仅条件温和,而且11-十一内酯类化合物和己内酯类化合物的收率高;环己酮类螺环过氧化物与氟醇溶剂的摩尔体积比为0.01mol/L~0.5mol/L,优选为0.04mol/L,反应时间为10min~60min,优选的时间为40min。
本发明中,反应结束后,可采用的后处理方式包括:加入无机碱(例如碳酸钾)中和质子酸,然后过滤、减压蒸馏可以得到11-十一内酯类化合物和己内酯类化合物。
2008年石井康敬、岩浜隆裕在专利CN 101627025A中采用六氟异丙醇作为溶剂,仲醇和酮同时作为底物,通过氧气氧化制备酯或内酯,并提出反应机理经过一种过氧化物中间体。在该专利工艺中,当采用六氟异丙醇或高氟叔丁醇为溶剂时,环己酮螺环过氧化物重排过程只能得到己内酯,而采用本发明中所述的氟醇溶剂时,可以同时得到11-十一内酯类化合物、己内酯类化合物和环癸烷类化合物。利用电子顺磁共振技术对反应机理进行分析,发现六氟异丙醇或高氟叔丁醇中可以检测到两种氧中心的自由基,而在本发明中所述的氟醇溶剂,例如2,2,2-三氟乙醇中可以检测到除上述两种自由基以外的第三种碳中心自由基,推测机理如Scheme 4所示,可以推断本发明中所采用的氟醇溶剂更有利于碳自由基中间体的产生,从而得到11-十一内酯类化合物与环癸烷类化合物。
Scheme 4氟醇溶剂中环己酮螺环过氧化物重排的可能反应机理
与现有的技术相比,本发明具有如下优点:
(1)本发明采用环己酮类螺环过氧化物为底物,相对于其他生产大环内酯的原料容易合成,价格低廉;
(2)本发明采用氟醇为溶剂,2,2,2-三氟乙醇为工业化应用非常广泛的一种氟醇溶剂,价格低廉,同时相对于之前的Story合成反应条件更为温和,操作安全且收率更高;
(3)本发明除了可以得到11-十一内酯,还可以得到己内酯,己内酯作为聚己内酯(PCL)单体也具有十分重要的工业价值。
具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于实施例表示的范畴。
实施例1
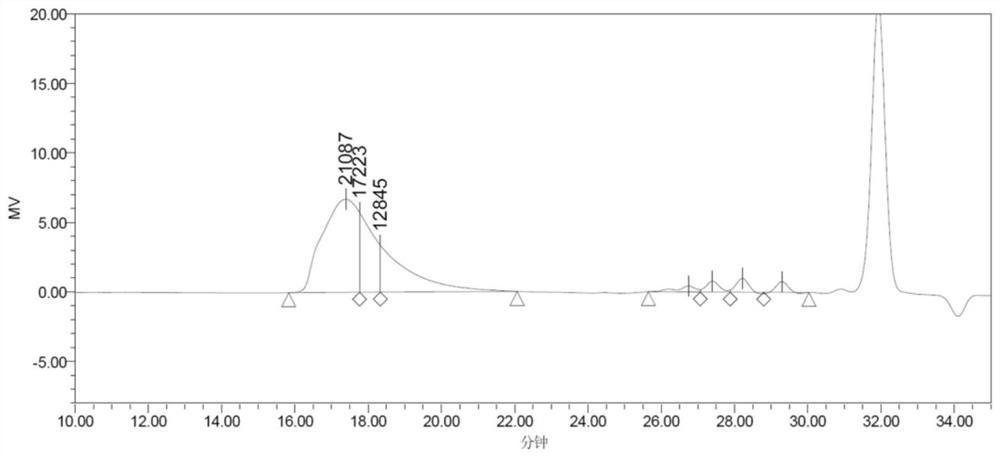
将50g环己酮螺环过氧化物,2g一水对甲基苯磺酸(p-TsOH·H2O)加入5000ml 2,2,2-三氟乙醇中,在25℃下机械搅拌40min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为43%,己内酯为32%,环癸烷为16%。
然后加入9g K2CO3继续搅拌5min。所得反应液过滤后在室温下旋蒸回收溶剂。所得混合物产品在0.03MPa下减压蒸馏,可得16.03g己内酯(收率为31.73%,GC纯度为99%),4.96g环癸烷(收率为15.18%,GC纯度为94%),18.05g 11-十一内酯(收率为42.50%,GC纯度为95%)。
实施例2
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml1,2,2-三氟乙醇中,在25℃下磁子搅拌40min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为35%,己内酯为33%,环癸烷为12%。
实施例3
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml1,1,2,2,2-五氟乙醇中,在25℃下磁子搅拌40min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为26%,己内酯为37%,环癸烷为10%。
实施例4
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2,2,3,3-四氟丙醇中,在25℃下磁子搅拌40min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为38%,己内酯为21%,环癸烷为8%。
实施例5
将0.05g环己酮螺环过氧化物,0.003g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌30min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为90%,产物选择性11-十一内酯为46%,己内酯为34%,环癸烷为16%。
实施例6
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2,2,2-三氟乙醇中,在45℃下磁子搅拌20min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为81%,产物选择性11-十一内酯为47%,己内酯为32%,环癸烷为14%。
实施例7
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2,2,2-三氟乙醇中,在60℃下磁子搅拌10min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为60%,产物选择性,11-十一内酯为50%,己内酯为35%,环癸烷为13%。
实施例8
将0.05g环己酮螺环过氧化物,0.001g质量浓度为38%的盐酸加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌1h。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为36%,己内酯为28%,环癸烷为12%。
实施例9
将0.05g环己酮螺环过氧化物,0.001g质量浓度为98%的硫酸加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌1h。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为34%,己内酯为26%,环癸烷为11%。
实施例10
将0.05g环己酮螺环过氧化物,0.001g三氟乙酸加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌1h。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为43%,己内酯为29%,环癸烷为13%。
实施例11
将0.05g环己酮螺环过氧化物,0.002g苯胂酸加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌2h。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为41%,己内酯为29%,环癸烷为14%。
实施例12
将0.05g环己酮螺环过氧化物,0.001g苯膦酸加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌1h。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为38%,己内酯为27%,环癸烷为11%。
实施例13
将0.058g对甲基环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌40min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性4,9-二甲基-11-十一内酯为43%,3-甲基-己内酯为31%,1,6-二甲基-环癸烷为18%。
实施例14
将0.062g对乙基环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌40min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性4,9-二乙基-11-十一内酯为40%,3-乙基-己内酯为29%,1,6-二乙基-环癸烷为13%。
实施例15
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入0.5ml 2,2,2-三氟乙醇中,在25℃下磁子搅拌40min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为31%,己内酯为20%,环癸烷为11%。
实施例16
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌60min。反应结束后,对反应混合物进行气相色谱分析,联苯作为内标,底物转化率为100%,产物选择性11-十一内酯为36%,己内酯为29%,环癸烷为16%。
对比例1说明了质子酸的催化作用
对比例1
将0.05g环己酮螺环过氧化物,直接加入5ml2,2,2-三氟乙醇中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
对比例2-9说明了氟醇溶剂的溶剂作用
对比例2
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml乙醇中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
对比例3
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2-氟乙醇中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
对比例4
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml2,2-二氟乙醇中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
对比例5
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml三氯乙醇中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
对比例6
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml全氟己烷中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
对比例7
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml全氟三乙胺中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
对比例8
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml全氟甲苯中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
对比例9
将0.05g环己酮螺环过氧化物,0.002g一水对甲基苯磺酸(p-TsOH·H2O)加入5ml全氟(2-丁基四氢呋喃)中,在25℃下磁子搅拌10h。对反应混合物进行气相色谱分析,联苯作为内标,环己酮螺环过氧化物转化率0%。
一种11-十一内酯类化合物和己内酯类化合物的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。





动态评分
0.0