

IPC分类号 : C07D473/00,C09K11/06,G01N21/64







专利摘要
本发明公开了基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂及其制备方法和应用,属于生物化学技术领域。本发明的基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂具有超大且可调的斯托克斯位移、可调的发射波长、较低的生物毒性、良好的光稳定性以及聚集诱导发光特性,将其与荧光传感技术联用实现了对脂滴的高灵敏度、高选择性、原位成像,同时该试剂合成路线简洁,反应条件温和,原料经济易得,为脂滴靶向染色荧光探针的设计提供了新思路与方法。
权利要求
1.一种基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂,其特征在于,其分子结构如(Ⅰ)式所示:
其中:
R
R
R
2.根据权利要求1所述的基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂,其特征在于,R
3.根据权利要求1所述的基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂,其特征在于,
R
R
R
4.根据权利要求1所述的基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂,其特征在于,
R
R
R
5.根据权利要求1所述的基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂,其特征在于,
R
R
R
6.根据权利要求1~5任一项所述的基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂的制备方法,其特征在于,包括以下步骤:
(1)将2,6-二氯嘌呤和R
(2)将所述第二中间体、强碱和有机溶剂混合搅拌,然后加入R
(3)将所述第二中间体、R
7.根据权利要求6所述的制备方法,其特征在于,
步骤(1)中,加热温度为50~120℃,搅拌时间为4~12h;
步骤(2)中,先后两次搅拌时间分别为1~2h和4~12h,加热温度为50~100℃;
步骤(3)中,回流时间为5~12h。
8.根据权利要求6或7所述的制备方法,其特征在于,
步骤(1)中,所述有机溶剂为DMSO和DM中的至少一种,所述弱碱为碳酸钠、碳酸钾、磷酸钾和磷酸钠中的至少一种;
步骤(2)中,所述有机溶剂为二氧六环和四氢呋喃中的至少一种,所述强碱为正丁基锂、叔丁醇钾、叔丁醇钠、氢化钾、氢化钠、碳酸钾和碳酸钠中的至少一种;
步骤(3)中,所述催化剂为四三苯基磷钯,所述混合溶剂为四氢呋喃与水的混合溶剂或者二氧六环与水的混合溶剂,所述弱碱为碳酸钾、碳酸钠、磷酸钾和磷酸钠中的至少一种。
9.根据权利要求1~5任一项所述的基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂在生物体脂滴上进行荧光成像中的用途。
说明书
技术领域
本发明涉及生物化学技术领域,具体涉及基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂及其制备方法和应用。
背景技术
脂滴,内部含有丰富的甘油三酯、固醇酯等中性脂质,是细胞内贮存能量的主要场所。此外,其内部的甾醇、脂肪酸和磷酸酯等,也为细胞内生物膜的形成提供了基础。因此,脂滴的多样性功能使得它在膜生物学、能量代谢起着重要作用,也使得脂滴成为维持细胞发展平衡的一个重要细胞器。然而,也正是由于其储存脂质的能力,近年来的病理学研究发现,脂滴的聚集与肥胖、糖尿病、动脉粥样硬化等疾病息息相关。综上所述,开发高选择性、高灵敏度的检测技术以准确对脂滴进行可视化,尤其是活体可视化,对于探索与解决医学早期诊断和研究生物学中的基本问题具有重要意义。
目前,对于观测脂滴的方法主要有普通光学显微镜观测、荧光染色标记、透射电镜、扫描电镜观测等方法。然而,由于脂滴本身近乎无色,故在普通光学显微镜、透射电镜、扫描电镜下不易区分脂滴。因此,现阶段已开发出一些用于脂滴染色的方法,使其更易于观察。苏丹Ⅲ、苏丹Ⅳ、油红O、锇酸等都是已经于普通光学显微镜观测中较为常用的手段,但这些染料均需使用固定后的细胞或组织、较大量的有机溶剂、繁琐的操作步骤,使得其难以在活体样品中进行长时间追踪性的观测,并不能满足现阶段研究的需求。近年来,荧光染色标记在脂滴研究领域发挥了重大作用。然而,现有的商售试剂BODIPY493/503,Nile Red等脂滴染料存在种类稀少、斯托克斯位移小、光稳定性较差等问题,因此开发新型的更具优势的脂滴染料显得尤为重要。
发明内容
为了解决现有脂滴染料斯托克斯位移小、光稳定性较差的问题,本发明的目的是提供一种基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂及其制备方法和应用。
本发明解决上述技术问题的技术方案如下:
一种基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂,其分子结构如(Ⅰ)式所示:
其中:
R1为脂溶性基团;
R2为 或者 其中Ar为芳香基团;
R3为脂溶性基团。
本发明以脂溶性取代基团作为活性基团,增强化合物的脂溶性能力以便于穿透细胞膜从而实现细胞内脂质存储亚细胞器到-脂滴-的靶向标记。
进一步地,在本发明较佳的实施例中,上述R1的脂溶性基团为C1-C20烷基、胆固醇或生物素;所述芳香基团为苯环、呋喃或者噻吩;R3的脂溶性基团为C1-C8烷基、吸电子基团或供电子基团,吸电子基团为 或者 供电子基团为 或者
本发明以C1-C20烷基、胆固醇或生物素作为R1基团,提高化合物脂溶性能力,以C1-C8烷基、吸电子基团或供电子基团为R3基团在提高化合物脂溶性能力的同时还调节荧光发射波段,实现化合物的荧光标记功能。
进一步地,在本发明较佳的实施例中,R1为C1-C10烷基;R2为 或者 Ar为苯环、呋喃或者噻吩;R3为C1-C8烷基、 或者
进一步地,在本发明较佳的实施例中,R1为 R2为 Ar为苯环、呋喃或者噻吩;R3为 或者
进一步地,在本发明较佳的实施例中,R1为 R2为 R3为 或者
基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂的制备方法,包括以下步骤:
(1)将2,6-二氯嘌呤和R1的卤代物R1-X溶于有机溶剂中,加入弱碱后加热搅拌,得到第一中间体;其中,X为Cl,Br或I;
(2)将第二中间体、强碱和有机溶剂混合搅拌,然后加入R2取代的 加热搅拌,得到第二中间体;
(3)将第二中间体、R3取代的 和催化剂溶于水和有机溶剂的混合溶剂中,加入弱碱回流,制得基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂。
进一步地,在本发明较佳的实施例中,步骤(1)中,加热温度为50~120℃,搅拌时间为4~12h;步骤(2)中,先后两次搅拌时间分别为1~2h和4~12h,加热温度为50~100℃;步骤(3)中,回流时间为5~12h。
进一步地,在本发明较佳的实施例中,步骤(1)中,有机溶剂为DMSO和DMF中的至少一种,弱碱为碳酸钠、碳酸钾、磷酸钾和磷酸钠中的至少一种;步骤(2)中,-有机溶剂为二氧六环和四氢呋喃中的至少一种,强碱为正丁基锂、叔丁醇钾、叔丁醇钠、氢化钾、氢化钠、碳酸钾和碳酸钠中的至少一种;步骤(3)中,催化剂为四三苯基磷钯,混合溶剂为四氢呋喃与水的混合溶剂或者二氧六环与水的混合溶剂,弱碱为碳酸钾、碳酸钠、磷酸钾和磷酸钠中的至少一种。
在本发明的合成过程中,第一步合成的底物2,6-二氯嘌呤由于溶解性较差,因而采用溶解能力良好的DMSO和DMF中的至少一种作为溶剂,并且由于烷基亲核较易发生且亲核试剂空间位阻小,因而添加碳酸钠、碳酸钾、磷酸钾和磷酸钠等弱碱促进反应进行;在第二步合成中,增加的烷基链能够增强化合物的脂溶性,因而采用二氧六环和四氢呋喃作为溶剂溶解,而且由于在第二步合成中引入了亲核较难发生且亲核剂空间位阻较大的芳香基团,因此采用正丁基锂、叔丁醇钾、叔丁醇钠、氢化钾、氢化钠、碳酸钾和碳酸钠等强碱促进反应进行。
基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂在生物体脂滴上进行荧光成像中的用途。
本发明具有以下有益效果:
本发明以嘌呤为基础的聚集诱导型染料改进,设计并合成了具有超大斯托克斯位移(>200nm)、高光稳定性的脂滴染料用于体外培养细胞、组织细胞的脂滴染色当中。本发明的染色试剂可以有效避免背景光的干扰,并可对脂滴进行较长时间的跟踪成像,提升了细胞成像结果的准确性,且染料几乎包含全光谱的发射范围,为实验研究提供了更多的选择性。此外,本发明提供的嘌呤骨架的聚集诱导型脂滴靶向染色试剂具有毒副作用小、原料经济易得、整条合成路线可操作性强、反应条件温和、总体成本较低等优势。并且,本发明提供的嘌呤骨架的聚集诱导型脂滴靶向染色试剂具有可调的斯托克斯位移、可调的发射波长、较低的生物毒性、良好的光稳定性以及聚集诱导发光特性,将其与荧光传感技术联用实现了对脂滴的高灵敏度、高选择性、原位成像,同时该试剂合成路线简洁,反应条件温和,原料经济易得,为脂滴靶向染色荧光探针的设计提供了新思路与方法。
附图说明
图1为本发明实施例制备方法的合成路线图。
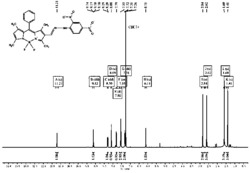
图2为实施例1的氢谱图。
图3为实施例2的氢谱图。
图4为实施例3的氢谱图。
图5为实施例4的氢谱图。
图6为实施例5的氢谱图。
图7为实施例6的氢谱图。
图8为实施例7的氢谱图。
图9为实施例8的氢谱图。
图10为实施例9的氢谱图。
图11为实施例10的氢谱图。
图12为实施例11的氢谱图。
图13为实施例1在DMSO溶液中的紫外吸收光谱图。
图14为实施例2在DMSO溶液中的紫外吸收光谱图。
图15为实施例3在DMSO溶液中的紫外吸收光谱图。
图16为实施例4在DMSO溶液中的紫外吸收光谱图。
图17为实施例5在DMSO溶液中的紫外吸收光谱图。
图18为实施例6在DMSO溶液中的紫外吸收光谱图。
图19为实施例7在DMSO溶液中的紫外吸收光谱图。
图20为实施例8在DMSO溶液中的紫外吸收光谱图。
图21为实施例9在DMSO溶液中的紫外吸收光谱图。
图22为实施例10在DMSO溶液中的紫外吸收光谱图。
图23为实施例11在DMSO溶液中的紫外吸收光谱图。
图24为实施例1在DMSO/H2O混合溶液中的发射光谱图。
图25为实施例2在DMSO/H2O混合溶液中的发射光谱图。
图26为实施例3在DMSO/H2O混合溶液中的发射光谱图。
图27为实施例4在DMSO/H2O混合溶液中的发射光谱图。
图28为实施例5在DMSO/H2O混合溶液中的发射光谱图。
图29为实施例6在DMSO/H2O混合溶液中的发射光谱图。
图30为实施例7在DMSO/H2O混合溶液中的发射光谱图。
图31为实施例8在DMSO/H2O混合溶液中的发射光谱图。
图32为实施例9在DMSO/H2O混合溶液中的发射光谱图。
图33为实施例10在DMSO/H2O混合溶液中的发射光谱图。
图34为实施例11在DMSO/H2O混合溶液中的发射光谱图。
图35为实施例1、2、3、4、5、6在DMSO/H2O混合溶液中的αAIE值。
图36为实施例7、8、9、10、11在DMSO/H2O混合溶液中的αAIE值。
图37为实施例1、2、3、4、5、6在H2O中的归一化荧光发射光谱图。
图38为实施例7、8、9、10、11在H2O中的归一化荧光发射光谱图。
图39为实施例1、2、3、4、5、6在固体状态下的归一化荧光发射光谱图。
图40为实施例1、2、3、4、5、6的MTS细胞毒性实验。
图41为实施例7、8、9、10、11的MTS细胞毒性实验。
图42为实施例1、2、3、4、5、6在HepG2细胞中的脂滴激光共聚焦实验。
图43为实施例7、8、9、10、11在HeLa细胞中的脂滴激光共聚焦实验。
具体实施方式
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
本发明实施例中,2,6-二氯嘌呤、苯硼酸、4-三氟甲基苯硼酸、4-氰基苯硼酸、4-甲酰基苯硼酸、丙二腈、1,3-茚二酮,各类溶剂、催化剂、碱购于伊诺凯科技有限公司,细胞株购于ATCC(American Type Culture Collection),10%胎牛血清(FBS)购于Hyclone,HepG2培养基购于美国Gibco。
请参照图1,本发明实施例基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂的制备方法其合成路线如图1所示,先采用2,6-二氯嘌呤和R1的卤代物R1-X在碱(特指上述提及的弱碱)和有机溶剂参与反应的条件下合成中间体1(也即是上述第一中间体),然后加入R2取代的 同样在碱和有机溶剂参与反应的条件下合成中间体2(第二中间体),最后加入R3取代的 在催化剂、弱碱和混合溶剂的作用下合成目标产物:基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂。
本发明基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂的制备方法,包括以下步骤:
(1)将2,6-二氯嘌呤和R1的卤代物R1-X溶于有机溶剂中,加入弱碱后在50~120℃下加热搅拌4~12h,得到第一中间体;其中,X为Cl,Br或I;有机溶剂为DMSO和DM中的至少一种,弱碱为碳酸钠、碳酸钾、磷酸钾和磷酸钠中的至少一种;
(2)将第二中间体、弱碱和有机溶剂混合搅拌1~2h,然后加入R2取代的 在50~100℃加热搅拌4~12h,得到第二中间体;有机溶剂为二氧六环和四氢呋喃中的至少一种,弱碱为正丁基锂、叔丁醇钾、叔丁醇钠、氢化钾、氢化钠、碳酸钾和碳酸钠中的至少一种;
(3)将第二中间体、R3取代的 和催化剂溶于水和有机溶剂的混合溶剂中,加入弱碱回流5~12h,制得基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂。催化剂为四三苯基磷钯,混合溶剂为四氢呋喃与水的混合溶剂或者二氧六环与水的混合溶剂,弱碱为碳酸钾、碳酸钠、磷酸钾和磷酸钠中的至少一种。
实施例1
本实施例的基于嘌呤骨架的聚集诱导型细胞膜靶向染色试剂的制备方法,包括以下步骤:
(1)合成中间体1:2,6-二氯-9-正丙基-9-氢-嘌呤(化合物1):
将2,6-二氯嘌呤(1.0mmol)、1-溴丙烷(1.5mol)和碳酸钾(3.0mmol)在DMSO(5mL)中混合搅拌6小时,随后加入100mL水。分离有机层,用乙酸乙酯(30mL×3)萃取水层。将有机萃取物用盐水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用石油醚/乙酸乙酯(3:2)洗脱,得到化合物1为白色固体,产率为57%。洗脱剂为乙酸乙酯/石油醚=2:3(V/V)。最终得白色固体,产率61%,其核磁共振氢谱数据如下:
(2)合成中间体2:2-氯-6-(1,2-二苯基吲哚)-9-正丙基-9-氢-嘌呤(化合物2):
在氮气保护下,将1,2-二苯基吲哚(3.76g,14mmol)加入到NaH(2g,21mmol,60%分散在矿物油中)在干燥THF(500mL)中的悬浮液中。将得到的溶液在0℃搅拌1小时,然后缓慢加入化合物1(3.2mL,14mmol,在50mL干THF中溶解)。将混合物加热至70摄氏度并搅拌过夜。随后加入水使反应猝灭。分离有机层,用乙酸乙酯(30mL×3)萃取水层。将有机萃取物用盐水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用石油醚/乙酸乙酯(3:1)洗脱,得到化合物2为白色固体,产率为54%,其核磁共振氢谱数据如下:
(3)合成基于基于嘌呤骨架的聚集诱导性脂滴靶向染色试剂:4-(6-(1,2-二苯基吲哚)-9-正丙基-9氢-嘌呤)-2-苯(化合物3)的合成:
于氮气保护下,将化合物2(509mg,1.1mmol)、苯硼酸(1.5eq)、四三苯基磷钯(0.05eq)和2.0mL碳酸钠水溶液(2M)加入10.0mL二氧六环中,回流8小时,薄层色谱监测反应完全后,将反应混合物倒入100mL水中,用二氯甲烷萃取。有机层用盐水、水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用二氯甲烷洗脱,得到化合物3为白色固体,产率为85%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C34H28N5
实施例2
6-(2,3-二苯基-1H-吲哚-1-基)-9-丙基-2-(4-(三氟甲基)苯基)-9H-嘌呤(化合物4)
本实施例与实施例1基本相同,区别在于改变化合物 中R3的取代基,其合成路线如下:
于氮气保护下,将化合物2(509mg,1.1mmol)、4-三氟甲基苯硼酸(1.5eq)、四三苯基磷钯(0.05eq)和2.0mL碳酸钠水溶液(2M)加入10.0mL二氧六环中,回流8小时,薄层色谱监测反应完全后,将反应混合物倒入100mL水中,用二氯甲烷萃取。有机层用盐水、水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用二氯甲烷洗脱,得到化合物4为白色固体,产率为81%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C35H26F3N5Na
实施例3
4-(6-(1,2-二苯基吲哚)-9-正丙基-9氢-嘌呤)-2-苯甲腈(化合物5)的合成:
本实施例与实施例1基本相同,区别在于改变化合物 中R3的取代基,其合成路线如下:
于氮气保护下,将化合物2(509mg,1.1mmol)、4-氰基苯硼酸(1.5eq)、四三苯基磷钯(0.05eq)和2.0mL碳酸钠水溶液(2M)加入10.0mL二氧六环中,回流8小时,薄层色谱监测反应完全后,将反应混合物倒入100mL水中,用二氯甲烷萃取。有机层用盐水、水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用二氯甲烷洗脱,得到化合物5为白色固体,产率为91%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C35H26N6Na
实施例4
4-(6-(1,2-二苯基吲哚)-9-正丙基-9氢-嘌呤)-2-苯甲醛(化合物6)的合成:
本实施例与实施例1基本相同,区别在于改变化合物 中R3的取代基,其合成路线如下:
于氮气保护下,将化合物2(509mg,1.1mmol)、4-甲酰基苯硼酸(1.5eq)、四三苯基磷钯(0.05eq)和2.0mL碳酸钠水溶液(2M)加入10.0mL二氧六环中,回流8小时,薄层色谱监测反应完全后,将反应混合物倒入100mL水中,用二氯甲烷萃取。有机层用盐水、水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用二氯甲烷洗脱,得到化合物6为黄绿色固体,产率为82%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C35H27N5O1Na
实施例5
2-(4-(6-(2,3-二苯基-1H-吲哚-1-基)-9-丙基-9H-嘌呤-2-基)亚苄基)丙二腈(化合物7)的合成:
本实施例与实施例4基本相同,区别在于最后引入脑文革缩合,其合成路线如下:
在乙醇(10mL)中加入化合物6(533mg,1mmol)和丙二腈(66mg,1mmol)后,将哌啶(0.05mL)滴入搅拌液中。然后将混合物在室温下搅拌约12小时,以薄层色谱监控反应完全后,旋蒸除去溶剂,用200-300目硅胶柱层析纯化。以二氯甲烷洗脱,得到深红色固体,产率为57%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C38H27N7Na
实施例6
2-(4-(6-(2,3-二苯基-1H-吲哚-1-基)-9-丙基-9H-嘌呤-2-基)亚苄基)丙二腈(化合物8)的合成:
本实施例与实施例4基本相同,区别在于最后引入脑文革缩合,其合成路线如下:
在乙醇(10mL)中加入化合物6(533mg,1mmol)和1,3-茚二酮(146mg,1mmol)后,将哌啶(0.05mL)滴入搅拌液中。然后将混合物在室温下搅拌约12小时,以薄层色谱监控反应完全后,旋蒸除去溶剂,用200-300目硅胶柱层析纯化。以二氯甲烷洗脱,得到橘黄色固体,产率为42%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C44H31N5O2Na
实施例7
4-(6-吲哚)-9-正丙基-9氢-嘌呤)-2-苯(化合物10)的合成:
(1)合成中间体1:2,6-二氯-9-正丙基-9-氢-嘌呤(化合物9)
在氮气保护下,将吲哚(1.64g,14mmol)加入到NaH(2g,21mmol,60%分散在矿物油中)在干燥THF(500mL)中的悬浮液中。将得到的溶液在0℃搅拌1小时,然后缓慢加入化合物1(3.2mL,14mmol,在50mL干THF中溶解)。将混合物加热至70摄氏度并搅拌过夜。随后加入水使反应猝灭。分离有机层,用乙酸乙酯(30mL×3)萃取水层。将有机萃取物用盐水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用石油醚/乙酸乙酯(3:1)洗脱,得到化合物2为白色固体,产率为57%,其核磁共振氢谱数据如下:
(2)合成基于基于嘌呤骨架的聚集诱导性脂滴靶向染色试剂:4-(6-吲哚)-9-正丙基-9氢-嘌呤)-2-苯(化合物10)的合成
于氮气保护下,将化合物9(343mg,1.1mmol)、苯硼酸(1.5eq)、四三苯基磷钯(0.05eq)和2.0mL碳酸钠水溶液(2M)加入10.0mL二氧六环中,回流8小时,薄层色谱监测反应完全后,将反应混合物倒入100mL水中,用二氯甲烷萃取。有机层用盐水、水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用二氯甲烷洗脱,得到化合物10为白色固体,产率为89%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C22H20N5
实施例8
4-(6-吲哚)-9-丙基-2-(4-(三氟甲基)苯基)-9H-嘌呤(化合物11)
本实施例与实施例7基本相同,区别在于改变第三中间体R3的取代基,其合成路线如下:
于氮气保护下,将化合物9(343mg,1.1mmol)、4-三氟甲基苯硼酸(1.5eq)、四三苯基磷钯(0.05eq)和2.0mL碳酸钠水溶液(2M)加入10.0mL二氧六环中,回流8小时,薄层色谱监测反应完全后,将反应混合物倒入100mL水中,用二氯甲烷萃取。有机层用盐水、水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用二氯甲烷洗脱,得到化合物11为白色固体,产率为91%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C22H19F3N5
实施例9
4-(6-吲哚)-9-正丙基-9氢-嘌呤)-2-苯甲腈(化合物12)的合成:
本实施例与实施例7基本相同,区别在于改变第三中间体R3的取代基,其合成路线如下:
于氮气保护下,将化合物9(343mg,1.1mmol)、4-氰基苯硼酸(1.5eq)、四三苯基磷钯(0.05eq)和2.0mL碳酸钠水溶液(2M)加入10.0mL二氧六环中,回流8小时,薄层色谱监测反应完全后,将反应混合物倒入100mL水中,用二氯甲烷萃取。有机层用盐水、水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用二氯甲烷洗脱,得到化合物12为白色固体,产率为91%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C23H19N6
实施例10
4-(6-吲哚)-9-正丙基-9氢-嘌呤)-2-苯甲醛(化合物13)的合成:
本实施例与实施例7基本相同,区别在于改变第三中间体R3的取代基,其合成路线如下:
于氮气保护下,将化合物9(343mg,1.1mmol)、4-甲酰基苯硼酸(1.5eq)、四三苯基磷钯(0.05eq)和2.0mL碳酸钠水溶液(2M)加入10.0mL二氧六环中,回流8小时,薄层色谱监测反应完全后,将反应混合物倒入100mL水中,用二氯甲烷萃取。有机层用盐水、水洗涤,并使用Na2SO4干燥。溶剂除减压蒸馏后,用200-300目硅胶柱层析纯化。用二氯甲烷洗脱,得到化合物13为黄绿色固体,产率为88%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C23H20N5O1
实施例11
2-(4-吲哚)-9-丙基-9H-嘌呤-2-基)亚苄基)丙二腈(化合物14)的合成:
在乙醇(10mL)中加入化合物13(381mg,1mmol)和丙二腈(66mg,1mmol)后,将哌啶(0.05mL)滴入搅拌液中。然后将混合物在室温下搅拌约12小时,以薄层色谱监控反应完全后,旋蒸除去溶剂,用200-300目硅胶柱层析纯化。以二氯甲烷洗脱,得到深红色固体,产率为90%,其核磁共振氢谱数据、碳谱数据和质谱数据分别如下:
HRMS(ESI):m/z:Calcd for C26H20N7
试验例1 吸光性验证
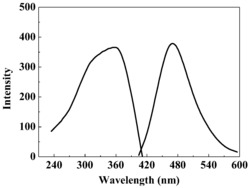
将上述化合物3、4、5、6、7、8、10、11、12、13、14配为5mM的DMSO母液。分别配为0.5、1、2、3、4、5、6、7、8、9、10uL的DMSO溶液,扫描紫外吸收值,绘图,检测结果如图13-图23所示,化合物3(图13),化合物4(图14),化合物5(图15),化合物6(图16),化合物7(图17),化合物8(图18),化合物10(图19),化合物11(图20),化合物12(图21),化合物13(图22),化合物14(图23)。从图13-图23可以看出,本发明实施例的制备的上述化合物3、4、5、6、7、8、10、11、12、13、14均表现出吸光性,能够吸收紫外光,且随着化合物浓度增加,对紫外光的吸收能力增加。
试验例2 聚集诱导特性表征
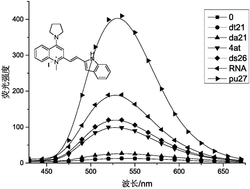
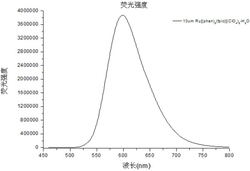
将上述化合物3、4、5、6、7、8、10、11、12、13、14配为5mM的DMSO母液。分别加入DMSO、水(H2O)和DMSO\H2O的混合溶液测定其荧光光谱,得到荧光发射曲线,如图24-图34所示。与DMSO溶液相比,在DMSO\H2O的混合溶液中,待测物的最大发射波长发生明显红移,荧光强度随H2O比例的增加逐渐增强,含H2O比例为100%时荧光强度达到最大。其中,化合物3(图24),化合物4(图25),化合物5(图26),化合物6(图27),化合物7(图28),化合物8(图29),化合物10(图30),化合物11(图31),化合物12(图32),化合物13(图33),化合物14(图34)。此外,图35为化合物3、4、5、6、7、8的αAIE值变化在DMSO/水中的荧光发射光谱汇总,图36为化合物10、11、12、13、14的αAIE值变化在DMSO/水中的荧光发射光谱汇总,在不良溶剂水含量增加的情况下,αAIE值的显著提升表明其优异的AIE特性。
图37为化合物3、4、5、6、7、8在水中的荧光发射光谱汇总,图38为化合物10、11、12、13、14在水中的荧光发射光谱汇总,图39为化合物3、4、5、6、7、8在固体状态下的荧光发射光谱汇总,这些数据表明系列探针实现了全光谱发光,提供了多种发射波段可以选择的空间。
试验例3 MTS细胞毒性实验
处于对数生长期的HepG2细胞接种于96孔培养板中,每孔接种10000个细胞,用含10%胎牛血清(FBS)、1%双抗的(青霉素-链霉素,1000KU/L)的DMEM培养基在37℃,5%CO2条件下培养过夜。待细胞完全贴壁,加入不同浓度梯度的化合物3、4、5、6、7、8、10、11、12、13、14,每个浓度设3个复孔,同时设空白对照组。加药后继续培养24小时,MTS法检测细胞的存活率,结果如图40、41所示。由此可说明,本发明实施例制得化合物对细胞毒害性小,能够适用于细胞脂滴显色成像。
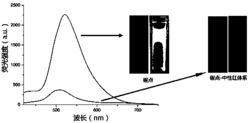
试验例4 对HepG2细胞(人肝癌细胞)进行脂滴染色的激光共聚焦成像
将约10000个HepG2细胞在35毫米培养皿中培养一夜。在DMSO<0.1vol%的1mL培养基中加入1μL 5mM的化合物母液,于37℃孵育30min,用PBS洗涤三次。在激光共聚焦显微镜下用适当的激发和发射滤光片对染料进行成像:λex=405nm,λem=450-600nm。结果如图42所示,探针与商售脂滴染料BODIPY荧光成像区域高度重叠,表明探针对于脂滴靶向的准确性。
试验例5 对HeLa细胞(宫颈癌细胞)进行脂滴染色的激光共聚焦成像
将约10000个HeLa细胞在35毫米培养皿中培养一夜。在DMSO<0.1vol%的1mL培养基中加入1μL 5mM的化合物母液,于37℃孵育30min,用PBS洗涤三次。在激光共聚焦显微镜下用适当的激发和发射滤光片对染料进行成像:λex=405nm,λem=450-600nm。结果如图43所示,探针与商售脂滴染料BODIPY荧光成像区域高度重叠,表明探针对于脂滴靶向的准确性。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
基于嘌呤骨架的聚集诱导型脂滴靶向染色试剂及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0