

IPC分类号 : C07J7/00,C07J17/00,C07J33/00,C07J71/00,A61K31/58,A61K31/57,A61P9/10,A61P9/04








专利摘要
达玛烷型三萜化合物及制备和应用。本发明属于药物化学、药理学与制剂领域,涉及达玛烷型三萜化合物治疗或预防心衰、心梗、冠心病、冠状动脉硬化、心肌缺血、心肌缺血再灌注的用途。
权利要求
1.一种达玛烷型三萜化合物,它的结构是如下任意一种:
2.根据权利要求1的达玛烷型三萜化合物在制备治疗或预防心肌缺氧再复氧损伤的药物中的应用。
说明书
技术领域
本发明属于药物化学、药理学与制剂领域,具体涉及达玛烷型三萜化合物化合物、制备、药理作用机制研究、应用、制剂,涉及达玛烷型三萜化合物治疗或预防心衰、心梗、冠心病、冠状动脉硬化、心肌缺血、心肌缺血再灌注的用途。
背景技术
目前,心血管疾病已经成为导致人类死亡的主要原因之一。心力衰竭是一种超负荷心脏病,是多种心血管及非心血管系统疾病的终末阶段,全世界约有260万人患有心力衰竭。该病的原发病多种多样,其发病机制亦错综复杂。
正性肌力药物是一类通过加强心肌收缩力而达到治疗心力衰竭作用的药物,主要包括以下几类:强心苷类,是一类Na+/K+-ATP酶抑制剂,如地高辛等;β肾上腺受体激动药,如多巴酚丁胺等;磷酸二酯酶抑制剂,如米力农等。然而,临床发现β肾上腺受体激动剂容易产生药物耐受性,PDE抑制剂容易加速疾病进程并增加死亡率。
Na+/K+-ATP酶是存在于细胞膜上的一种膜蛋白,能将3个钠离子泵出细胞外的同时将2个钾离子泵入细胞内。当Na+/K+-ATP酶被抑制后,细胞内钠离子逐渐增加,通过细胞膜上Na+-Ca2+交换系统,使细胞内钙离子浓度升高,进而达到正性肌力作用。因此,Na+/K+-ATPATP酶是一个非常重要的且明确的抗心力衰竭的作用靶标。
人参三醇等达玛烷型五环三萜类化合物具有降血糖、抗肿瘤、抗炎、降低胆固醇等多方面具有广泛的生物活性,并且具有中等强度的抗心肌缺血再灌注活性。因此,对人参三醇进行结构修饰提高其药理活性,对发现能够应用于市场的先导化合物具有重大的意义。
本发明主要对于人参三醇C-2,C-3、C-20,C-22位进行结构修饰,通过氧化,酯缩合、O3氧化、水解等一系列反应进行结构修饰,通过体外分子水平、细胞水平以及体内动物水平研究,发现具有显著抗心力衰竭活性的化合物。
技术方案
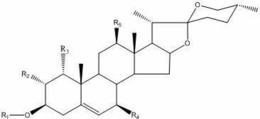
本发明涉及达玛烷型三萜化合物,结构通式为:
R1为H时,R2,R3选自 中的任意一种,
或R2选自H、=NOH中的任意一种,
或:R3选自=O、-OH、-OBn、=NOH、 中的任意一种;
R1,R2为 时,R3选自=O;
或:R4选自=O、-OH、-OAc、-OBn中的任意一种,
或:R5选自 中的任意一种。
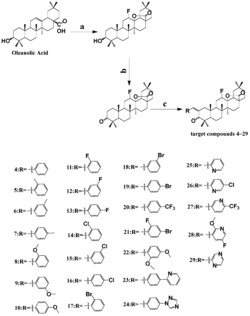
本发明涉及达玛烷型三萜化合物,它的结构是如下任意一种或者多种:
本发明的达玛烷型三萜化合物用于制备治疗或预防心梗的组合物。
本发明的达玛烷型三萜化合物用于制备治或预防疗心衰的组合物。
本发明的达玛烷型三萜化合物用于制备治或预防疗心肌缺血的组合物。
本发明的达玛烷型三萜化合物用于制备治疗或预防冠心病的组合物。
本发明的达玛烷型三萜化合物用于制备治疗或预防冠状动脉硬化的组合物。
本发明的达玛烷型三萜化合物用于制备治疗或预防心肌缺血再灌注所涉及的疾病的组合物。
本发明的达玛烷型三萜化合物的组合物,组合物含有0.01-500mg的达玛烷型三萜化合物。
本发明的达玛烷型三萜化合物的组合物,每制剂单位含有0.1-200mg的达玛烷型三萜化合物。
本发明涉及达玛烷型三萜化合物的组合物,每制剂单位含有1-100mg的达玛烷型三萜化合物。
本发明的达玛烷型三萜化合物:其特征在于:采用本领域的辅料制备得到药物、保健品。
本发明的达玛烷型三萜化合物的组合物,包括胶囊剂、片剂、颗粒剂、微丸、缓释胶囊剂、冻干粉、注射剂等。
本发明的药效学实验
药效学实验一:化合物对Na+/K+-ATP酶抑制作用的体外研究
一、材料与方法
1.实验材料
新合成化合物由北京大学医学部提供。人源Na+/K+-ATPase(Yanjing Co.,Ltd,Shanghai,China);ATP(MP Biomedicals,Irvine,CA,USA)。
2.实验仪器
FORMA 700型超低温冰箱,Thermo公司;YC-300L型药品储存柜,中科美菱低温科技有限责任公司;Direct-Q with pump型超纯水仪,Millopore公司;SW-CJ-2FD型超净化工作台:苏州净化设备有限公司;Forma 3111型水套式CO2培养箱:Thermo Electron company;Berthold LB941微孔板式多功能酶标仪,Berthold公司。
3.实验方法
(1)实验分组情况及药物浓度选择
所有化合物浓度选择:0μM、0.01μM、0.1μM、0.5μM、1μM、5μM。化合物均用DMSO助溶,0μM为含等体积的DMSO作为空白对照组,DMSO的终浓度为1%(v/v)。
(2)Cell free系统下酶活性检测
将浓度(0μM、0.01μM、0.1μM、0.5μM、1μM、5μM)的不同化合物与0.03U/ml Na+/K+-ATPase加至含Na+125mM,K+2.8mM,Mg2+4.5mM,EDTA 0.5mM的Tris(24mM,pH 7.8)缓冲液中,混合后在37℃孵育15分钟,再加入ATP 5mM检测15分钟内ATP释放的无机磷酸盐含量。Taussky-Shorr染色后,酶标仪检测。设置空白孔后,立即记录655nm处吸光度。GraphPad Prism软件拟合抑制曲线并计算IC50值。抑制率=(对照组OD值-实验组OD值)/对照组OD值×100%。
二、实验结果
表1.不同化合物对Na~+K~+atp酶的体外IC50值
实验结果显示:本发明对一系列达玛烷型三萜衍生物进行了体外人源的Na+/K+-ATPase抑制活性评价,结果显示化合物1-10对Na+/K+-ATPase具有显著抑制活性,与阳性对照化合物Digoxin的抑制活性相当。
药效学实验二:化合物对缺氧再复氧模型介导的心肌细胞损伤实验
一、材料与方法
1.实验材料
新合成化合物由北京大学医学部提供。连二亚硫酸钠(Sigma-Aldrich,St.Louis,MO);无糖DMEM培养基(Gibco,Grand Island,NY,USA);葡萄糖(Glucose,Glu)(国药集团化学试剂有限公司);青霉素(Sigma-Aldrich,St.Louis,MO);链霉素(Sigma-Aldrich,St.Louis,MO);胰蛋白酶(Gibco,Grand Island,NY,USA);胎牛血清(Fetal bovine serum,FBS)(Hyclone,Logan,UT,USA)。丁苯酞(Sigma-Aldrich,St.Louis,MO)。
清洁级SD孕大鼠,体重200–220g,雌性,购自北京维通利华实验动物技术有限公司,饲养于SPF级动物房,室内温度控制在23±2℃,自由饮食和摄水,昼夜时间为12h/12h。
2.实验仪器
三气培养箱,Thermo公司;SHZ-III型循环式水泵,南京科尔仪器设备有限公司;Direct-Q with pump型超纯水仪,Millopore公司;3K15型低温高速离心机,Sigma公司;TCL-16G-A型高速冷冻离心机:上海安亭科学仪器厂;Forma 3111型水套式CO2培养箱:Thermo Electron company;YXQ-LS-50SⅡ型立式压力蒸汽灭菌器;Berthold LB941微孔板式多功能酶标仪,Berthold公司。
3.实验方法
(1)药物浓度设置及分组:
人参皂苷元衍生物及丁苯酞(NBP)按照相应要求配置成母液,按照倍数稀释后用于后续实验。空白对照组(原代心肌细胞+完全DMEM培养基培养50h);OGD/R实验组(原代心肌细胞+浓度为(0μM,0.1μM,1μM,10μM)的衍生物培养24h后,缺氧2h,复氧24h);NBP阳性药物组(原代心肌细胞+浓度为(0μM,0.1μM,1μM,10μM)的NBP培养24h后,缺氧2h,复氧24h)。
(2)实验步骤:
大鼠心肌细胞的原代培养:取SD大鼠乳鼠(0-1d),75%乙醇溶液消毒,无菌台内取乳鼠心肌组织剪碎,加入0.125%胰蛋白酶与1%II型胶原酶混合液于37℃水浴振荡器中消化10min,用含10%胎牛血清(FBS)的DMEM终止消化,吸取混合液。重复消化5次,收集消化液,然后用200目筛网过滤,滤液离心(800rpm,10min),弃去上清液,加适量培养液悬浮沉淀,接种于6孔塑料培养板,置37℃、5%CO2孵箱中培养。
大鼠心肌细胞缺氧缺糖损伤模型:选取生长到第7d左右的细胞进行体外OGD/R模型。调节细胞密度,以4×104的细胞密度接种于48孔培养板中。除空白对照组外,其余使用分别含有浓度为(0,0.1μM,1μM,10μM)的wq类药物或NBP以及10%FBS含糖的DMEM培养基,培养24h后开始制备OGD/R模型。首先将培养基更换为不含葡萄糖与血清DMEM培养基,同时将细胞置于含有5%CO2、95%N2三气培养箱中培养2h,完成缺氧和糖的过程;随后将细胞培养基更换为完全含10%FBS及含糖的DMEM培养基,同时将细胞置于含有5%CO2、20%O2培养箱中培养24h,完成复氧过程,复氧结束前4h,每孔加入20μL MTT溶液(5mg/mL)。孵育结束后,弃去各孔上清液,每孔加入150μL DMSO,细胞振荡仪上振荡10min,待结晶物充分溶解后用酶标仪测定OD570。
(3)数据统计
通过设置模型对照组,计算不同组别原代心肌细胞存活率,结果采用mean±SD形式表示。数据组间统计学差异采用two-way ANOVA和Tukey’s检验,P值小于0.05认为有显著性差异。公式:survival rate=(实验组OD值/control组OD值)×100%。
二、实验结果
表2化合物对于大鼠心肌细胞存活率的影响.
实验结果显示:本发明对一系列达玛烷型三萜衍生物进行了体外大鼠心肌细胞缺氧再复氧模型介导的损伤实验,结果显示化合物1-10对缺氧再复氧心肌细胞损伤具有显著的保护作用。
药效学实验三:化合物3对大鼠体内心肌缺血再灌注的保护作用
一、材料与方法
1.实验材料
SPF级SD大鼠,220g-240g。购自北京维通利华实验动物技术有限公司。谷草转氨酶(AST/GOT)试剂盒、肌酸激酶同工酶(CK-MB)试剂盒购自南京建成生物工程研究所。
2.实验仪器
GZX-9140MBE型鼓风干燥箱,上海博迅实业有限公司医疗设备厂;Direct-Q with pump型超纯水仪,Millopore公司;BS224型电子天平:北京塞多利斯仪器系统有限公司;3K15型低温高速离心机,Sigma公司;TCL-16G-A型高速冷冻离心机:上海安亭科学仪器厂;SW-CJ-2FD型超净化工作台:苏州净化设备有限公司;YXQ-LS-50SⅡ型立式压力蒸汽灭菌器;Berthold LB941微孔板式多功能酶标仪,Berthold公司。
3.实验方法
(1)药物剂量设置及分组:
将SD大鼠分为8组,每组5只(n=5):1)假手术组(Sham):开胸,但不结扎冠状动脉,造模前灌胃给药40mg/kg生理盐水三天,早晚各一次。2)单加药组(D):开胸,但不结扎冠状动脉,造模前灌胃给药40mg/kg化合物3三天,早晚各一次。3)模型组(IR):结扎冠状动脉左前降支,再灌注24h。造模前灌胃给药40mg/kg生理盐水三天,早晚各一次。4)高剂量组(IR+40mg/kg):结扎冠状动脉左前降支,再灌注24h。造模前灌胃给药40mg/kg化合物3三天,早晚各一次。5)中剂量组(IR+20mg/kg):结扎冠状动脉左前降支,再灌注24h。造模前灌胃给药20mg/kg化合物3三天,早晚各一次。6)低剂量组(IR+10mg/kg):结扎冠状动脉左前降支,再灌注24h。造模前灌胃给药10mg/kg化合物3三天,早晚各一次。7)阳性对照组(IR+卡托普利):结扎冠状动脉左前降支,再灌注24h。造模前灌胃给药20mg/kg卡托普利三天,早晚各一次。8)人参三醇对照组(IR+人参三醇):结扎冠状动脉左前降支,再灌注24h。造模前灌胃给药40mg/kg人参三醇三天,早晚各一次。
(2)大鼠心肌缺血再灌注模型的制备
大鼠心肌缺血再灌注损伤模型制备,用3%戊巴比妥钠溶液(0.2ml/100g)对大鼠进行腹腔麻醉,仰卧位固定四肢于手术台上,连接肢体心电图电极,描记标准16导联心电图。经口气管插管,连接小动物呼吸机,潮气量调整至15mL/kg,呼吸频率70次/min,呼吸比1:1.5。前胸脱毛,于低3-4助间开胸逐层钝性分离筋膜肌肉,暴露心脏,撕开心包膜,于左心耳下方1-2mm出进针,横跨3-4mm,进针深度约为1.5-2mm,用6-0号缝线推管法结扎冠状动脉左前降支。心电图实时监测并记录,以ST段持续抬高,结扎点远端供血心肌逐渐紫绀,苍白说明心肌缺血造模成功;30min后解除结扎线,缝合,以ST段下降及缺血苍白的心肌变红润作为再灌注模型成功的标志,所有动物在再灌注24h后,腹主动脉取血,3500rpm,10min分离血浆,-80℃保存备用;取下心脏,-80℃保存备用。
(3)Evans Blue/TTC检测心肌梗死面积
再灌注24h后,麻醉大鼠(方法同造模),腹主动脉取血,将心脏取出,以生理盐水冲洗,放入-80℃中7min后取出,由心尖处,沿结扎线方向,将心脏横切为1-2mm后的薄片5-7个。放入1%的TTC溶液中,37℃水浴加热12min,放入中性福尔马林中固定,室温下静置过夜,次日,拍照,用Image-Pro plus5.0图像分析软件测量每个薄片心肌梗死面积(心肌梗死比例=梗死区面积/左心面积)。所有动物在再灌注24h后,腹主动脉取血,血样置于离心管中3500rpm离心10min分离血浆,-80℃保存备用。检测方法按照相应测定试剂盒说明书进行。
二、实验结果
实验结果显示:化合物3在高中低剂量均可显著减少梗死面积,呈现浓度依赖性,并且在高中低剂量减少心肌梗死面积的作用均强于阳性药卡托普利。此外在给药剂量为40mg/kg时,化合物3减少心肌梗死面积的作用强于母体化合物人参三醇。
实验结果显示:与假手术组和单加药组比较,再灌注模型组血清中的AST、CK活性均显著增加;与缺血再灌注模型组比较,化合物3高、中、低三个剂量组都能明显降低血清中AST和CK活性。
附图说明
图1:化合物3对大鼠缺血再灌注心肌梗死面积的影响
图2:化合物3对缺血再灌注大鼠血浆肌酸激酶(CK)和天冬氨酸转氨酶(AST)水平的影响
具体实施例
实施例1
化合物12、13的制备
将476mg化合物11即20(R)-人参三醇溶解于30mL二氯甲烷中,加入戴斯马丁氧化剂424mg,室温下反应3小时。反应结束后,用饱和碳酸氢钠和亚硫酸氢钠淬灭反应。二氯甲烷萃取,减压回收溶剂后,得产物粗品。硅胶柱层析(石油醚/乙酸乙酯=5:3)得化合物12和化合物13。化合物12:13C NMR(100MHz,CDCl3):δ211.90,78.51,76.63,73.30,69.43,65.62,54.83,52.65,51.08,50.20,48.95,46.29,43.65,39.94,37.58,36.45,35.71,33.06,31.14,30.85,27.56,27.16,27.05,24.90,19.44,17.33,17.05,16.27,15.68,15.16.ESI-MS:m/z 473.68[M-H]-。化合物13:13C NMR(100MHz,CDCl3):δ219.33,76.73,73.35,69.75,67.87,59.07,54.66,51.18,48.97,48.37,47.29,45.21,40.50,39.78,38.14,36.52,35.81,33.12(2C),32.05,31.35,31.25,27.23,25.18,19.64,19.52,17.68,17.04,16.35,16.03.ESI-MS:m/z 473.68[M-H]-.
实施例2
化合物1的制备
将284mg化合物13溶于20mL四氢呋喃,加入120mg NaH,反应1小时。加入205mg溴化苄,加热至70℃下反应8小时。反应结束后,乙酸乙酯萃取,减压蒸干溶剂得粗产品。硅胶柱层析(石油醚/乙酸乙酯=6:1)得化合物1。
1H NMR(400MHz,CDCl3):7.48-7.16(m,5H),6.48(s,1H),4.02(m,1H),3.49(m,1H),3.19(m,1H),3.06(m,1H),2.40(m,1H),1.35(s,3H),1.33(s,3H),1.28(s,3H),1.23(s,3H),1.19(s,3H),1.02(s,3H),0.90(s,3H),0.78(s,3H).13C NMR(100MHz,CDCl3):δ219.12,140.53,129.04(2C),128.55(2C),126.05,76.84,73.45,69.75,67.97,58.35,54.66,51.18,49.00,48.91,48.73,47.52,45.12,42.92,40.47,38.17,36.87,36.52,35.82,33.13,32.15,31.26(2C),27.26,25.19,19.53,19.21,19.18,17.02,16.36,16.00.ESI-MS:m/z 565.84[M+H]+。
实施例3
化合物2的制备
将474mg化合物2溶于30mL四氢呋喃,加入200mg NaH反应1小时。加入0.24mL溴化苄,70℃下反应8小时。反应结束后,乙酸乙酯萃取,减压蒸干溶剂得粗产品。硅胶柱层析(石油醚/乙酸乙酯=6:1),得化合物2。1H NMR(400MHz,CDCl3):δ7.33-7.21(m,5H),6.32(s,1H),4.63(d,J=12.0Hz,1H),4.40(d,J=12.0Hz,1H),3.60(td,J=10.3Hz,4.8Hz,1H),2.82(dd,J=11.5Hz,3.6Hz,1H),2.53(d,J=11.6Hz,1H),2.09(s,1H),1.25(s,6H),1.21(s,3H),1.16(s,3H),1.03(s,3H),0.97(s,6H),0.91(s,3H).13C NMR(100MHz,CDCl3):δ211.58,139.17,128.17(2C),127.39(2C),127.25,85.71,76.55,73.19,71.25,69.35,65.91,54.77,52.60,50.98,50.13,48.90,46.18,43.48,39.79,37.74,36.37,35.63,33.01,31.05,30.85,27.75,27.09,24.83,22.69,19.37,17.26,17.00,16.23,16.21,15.60.ESI-HRMS(m/z)[M+H]+calcd for C37H57O4,565.4257;found,565.4246.
实施例4
化合物14的制备
将379mg化合物13溶于30mL无水四氢呋喃中,加入346mg无水甲醇钠,0.87mL草酸二乙酯,室温下反应6小时。反应结束后,乙酸乙酯萃取,减压蒸干溶剂得粗产品。将粗产品溶解于30mL乙醇中,加入110mg盐酸羟胺,加热回流反应4h。反应停止后,乙酸乙酯萃取,减压蒸干得到粗产品,经柱层析纯化(石油醚/乙酸乙酯=2/1)后,得化合物14。1H NMR(400MHz,CDCl3):δ6.50(s,1H),4.41(q,J=7.1Hz,2H),4.14(m,1H),3.61(td,J=10.0Hz,4.7Hz),2.89(d,J=16.0Hz,1H),2.13(d,J=16.0Hz,1H),1.58(s,3H),1.51(s,3H),1.39(t,J=7.2Hz,3H),1.28(s,3H),1.23(s,3H),1.20(s,3H),1.09(s,3H),0.93(s,3H),0.85(s,3H).13C NMR(100MHz,CDCl3):δ177.15,160.78(2C),110.26,76.76,73.36,69.68,67.80,61.76,59.13,54.76,51.20,48.98,47.85,46.00,40.74,40.57,36.57(2C),36.22,35.86,33.12,32.56,31.32,31.00,27.35,25.22,20.68,19.57,17.12,17.10,16.70,16.37,14.39.ESI-HRMS(m/z)[M+H]+calcd for C34H54NO6,572.3951;found,572.3952。
实施例5
化合物3的制备
将化合物258mg化合物14溶于20mL乙醇中,加入氢氧化锂34mg,室温,反应12小时。反应结束后,加入1M的盐酸溶液,乙酸乙酯萃取,减压蒸干溶剂得粗产品。硅胶柱层析(二氯甲烷/甲醇=20/1),得到化合物3。1H NMR(400MHz,CD3OD):δ4.08(m,1H),3.62(m,1H),2.92(d,J=15.8Hz,1H),2.18(d,J=15.8Hz,1H),2.03-1.96(m,2H),1.59(s,3H),1.52(s,3H),1.29(s,3H),1.22(s,6H),1.11(s,3H),0.98(s,3H),0.88(s,3H).13C NMR(100MHz,CD3OD):δ178.60,163.08,155.82,111.57,78.17,74.61,71.39,67.85,59.73,55.93,52.31,49.84,48.73,46.01,41.55,41.39,37.41(2C),37.29,36.75,33.45,32.79,32.23,31.62,27.66,26.08,20.94,19.89,17.49,17.33,17.12,16.86.ESI-HRMS(m/z)[M+H]+calcd forC32H50NO6,544.3638;found,544.3639。
实施例6
化合物4的制备
将474mg化合物13溶于乙醇中,加入139mg盐酸羟胺,168mg碳酸氢钠,加热回流2小时。反应结束待冷却至室温后,以二氯甲烷萃取。减压蒸干溶剂得粗产品。硅胶柱层析(石油醚/乙酸乙酯=2:1)得化合物4。1H NMR(400MHz,CDCl3):δ6.35(s,1H),4.02(m,1H),3.50(m,1H),3.15(m,1H),2.10-2.01(m,1H),1.96-1.89(m,1H),1.39(s,3H),1.37(s,3H),1.24(s,3H),1.19(s,3H),1.16(s,3H),1.00(s,3H),0.88(s,3H),0.82(s,3H).13C NMR(100MHz,CDCl3):δ167.82,76.72,73.30,69.85,67.75,58.36,54.62,51.16,48.94,48.79,45.49,40.40,40.39,38.75,38.47,36.52,35.80,33.78,33.11,31.31,31.21,27.21,25.17,21.03,19.49,18.02,17.02,16.96,16.34,16.12.ESI-HRMS(m/z)[M+H]+calcd for C30H52NO4,490.3896;found,490.3894。
实施例7
化合物5的制备
将474mg化合物13溶于20mL叔丁醇,加入672mg叔丁醇钾,缓慢加入0.4mL亚硝酸异戊酯,室温下反应3小时。反应结束后,二氯甲烷萃取。有机层减压蒸干溶剂得粗产品。硅胶柱层析(石油醚/甲乙酸乙酯=1:1),得化合物5。1H NMR(400MHz,CD3OD):δ3.96(m,1H),3.61(m,1H),2.90(d,J=18.6Hz,1H),2.32(d,J=18.6Hz,1H),2.04-1.97(m,1H),1.92-1.82(m,3H),1.45(s,3H),1.29(s,6H),1.22(s,6H),1.09(s,3H),0.98(s,3H),0.87(s,3H).13C NMR(100MHz,CD3OD):δ207.00,155.18,78.18,74.63,71.31,67.63,59.29,55.90,52.29,49.88,47.92,47.85,45.26,43.35,41.12,37.69,37.40,36.74,33.46,32.25,32.16,31.99,27.64,26.06,20.67,19.88,17.87,17.26,17.13,16.12.ESI-HRMS(m/z)[M+H]+calcd for C30H50NO5,504.3689;found,504.3676。
实施例8
化合物6的制备
将503mg化合物5溶于10mL无水吡啶中,加入0.21m乙酰氯,加热回流2小时。反应结束后,加1M的稀盐酸,乙酸乙酯萃取。有机层减压蒸干溶剂得粗产品。硅胶柱层析(石油醚/甲乙酸乙酯=2:1),得化合物6。1H NMR(400MHz,CDCl3):δ6.34(s,1H),5.34(m,1H),3.68(m,1H),2.79(m,1H),2.49(s,3H),2.21-2.14(m,2H),2.09(s,3H),1.44(s,3H),1.35(s,3H),1.27(s,3H),1.23(s,3H),1.22(s,3H),1.20(s,6H),0.91(s,3H).13C NMR(100MHz,CDCl3):δ195.73,171.60,170.08,161.48,131.07,76.75,73.38,69.76,69.46,57.19,54.68,51.08,48.63,47.12,42.96,42.48,41.72,41.16,36.53,35.80,33.21,32.68,31.33,31.26,27.29,25.02,21.90,21.57,20.72,19.53,17.39,16.92,16.32,14.30.ESI-HRMS(m/z)[M+H]+calcd for C34H52NO6,570.3795;found,570.3802。
实施例9
化合物15的制备
将238mg人参三醇化合物11溶于30mL二氯甲烷,加入324mg氯铬酸吡啶鎓盐,室温下反应3小时。反应结束后过滤,滤液减压蒸去溶剂,得到粗产品。硅胶柱层析(石油醚/乙酸乙酯=6:1),得化合物15。13C NMR(100MHz,CDCl3):δ214.44,210.97,76.57,73.30,69.20,65.30,54.79,52.45,51.01,49.78,48.94,46.95,46.16,43.38,41.14,36.38,35.64,33.90,33.02,31.12,30.93,27.13,24.83,24.18,21.63,19.40,17.25,16.42,16.22,15.75.ESI-MS:m/z 471.65[M-H]-.
实施例10
化合物7的制备
将236mg化合物7溶于10mL冰醋酸中,加入131mg的4-甲氧基苯肼盐酸盐,加热回流4小时。反应结束后出去过量的酸,乙酸乙酯萃取,有机层减压蒸干溶剂得粗产品。硅胶柱层析(石油醚/乙酸乙酯=8:1),得化合物7。1HNMR(400MHz,CDCl3):δ7.78(s,1H),7.19(d,J=8.7Hz,1H),6.84(br s,1H),6.78(dd,J=8.7Hz,1.8Hz,1H),6.49(s,1H),3.84(s,3H),3.74(td,J=10.3Hz,4.8Hz,1H),2.96(d,J=15.0Hz,1H),2.67(s,1H),2.64(d,J=12.3Hz,1H),2.57(d,J=15.0Hz,1H),2.34-2.24(m,2H),2.01(d,J=12.3Hz,1H),1.72(s,3H),1.39(s,3H),1.29(s,3H),1.25(s,3H),1.22(s,3H),1.07(s,3H),1.06(s,3H),1.03(s,3H).13C NMR(100MHz,CDCl3):δ212.06,153.99,142.14,131.34,128.29,111.35,111.28,105.38,100.14,76.73,73.45,69.55,64.15,55.97,54.82,52.62,51.17,49.25,49.22,45.18,44.24,39.53,36.53,35.81,34.37,33.09,31.77,31.35,30.77,27.26,24.98,24.59,19.57,18.65,17.39,16.37,15.50.ESI-HRMS(m/z)[M+H]+calcd for C37H54NO4,576.4053;found,576.4055。
实施例11
化合物8的制备
将472mg化合物15溶于250mL四氢呋喃,在冰水浴条件下,缓慢加入三溴化吡啶鎓480mg,常温下反应12h。硫代硫酸钠淬灭反应,乙酸乙酯萃取。减压蒸干有机溶剂得粗产品。粗产品溶于30mL无水乙醇中,加入532mg硫脲,加热回流反应4小时。反应停止后冷却至室温,加水稀释,乙酸乙酯萃取,有机相减压蒸干得粗产品。硅胶柱层析(石油醚/乙酸乙酯=8/1),得化合物8。1H NMR(400MHz,CDCl3):δ6.38(s,1H),4.38(q,J=6.8Hz,2H),3.61(m,1H),3.01(d,J=16.6Hz,1H),2.70(d,J=16.6Hz,1H),2.58-2.56(m,2H),2.20(br d,1H),2.00-1.90(m,3H),1.68(s,3H),1.40(s,3H),1.22(s,3H),1.18(s,3H),1.14(s,3H),0.99(s,3H),0.97(s,3H),0.95(s,3H).13C NMR(100MHz,CDCl3):δ210.37,160.67,160.13,155.14,132.22,76.57,73.34,69.15,63.09,62.23,54.69,52.25,50.96,48.97,48.73,44.89,43.98,41.12,37.40,36.39,35.66,33.01,31.51,31.22,30.63,27.12,24.84,23.99,19.44,18.40,17.19,16.22,15.31,14.33.ESI-HRMS(m/z)[M+H]+calcd for C34H52NO5S,586.3566;found,586.3570。
实施例12:
化合物16的合成
将472mg化合物7溶于30mL乙醇,加152mg盐酸羟胺和202mg碳酸氢钠,加热回流2小时。反应结束加水稀释,以二氯甲烷萃取。有机层减压蒸干得粗产品,硅胶柱层析(石油醚/乙酸乙酯=4:1),得化合物16。1H NMR(400MHz,CDCl3):δ6.38(s,1H),3.57(m,1H),3.26(m,1H),2.51(d,J=11.5Hz,1H),2.31(s,1H),2.02-1.87(m,5H),1.81(d,J=11.5Hz,1H),1.77-1.69(m,2H),1.41(s,3H),1.22(s,3H),1.17(s,3H),1.13(s,6H),1.02(s,3H),0.96(s,3H),0.92(s,3H).13C NMR(100MHz,CDCl3):δ211.77,165.21,76.62,73.33,69.37,66.19,54.83,52.64,51.04,50.03,48.92,46.33,43.79,40.27,39.98,36.43,35.69,33.06,31.10,30.84,27.15,25.35,24.86,22.84,19.41,17.21,16.69,16.44,16.25,15.69.ESI-MS:m/z 488.41[M+H]+。
实施例13
化合物9的制备
将487mg化合物16溶于THF,加入80mg的NaH,常温下反应1小时。加入0.24mL 2-氯-5-(氯甲基)噻吩,60℃下反应8小时。加水淬灭反应,乙酸乙酯萃取,有机层减压蒸干溶剂得粗产品。硅胶柱层析(石油醚/乙酸乙酯=10:1),得到化合物9。1H NMR(400MHz,CDCl3):δ6.77-6.74(m,2H),6.38(s,1H),5.06-5.4.98(m,2H),3.56(m,1H),2.98(m,1H),2.50(d,J=11.9Hz,1H),2.22(s,1H),2.14-2.07(m,1H),1.28(s,3H),1.26(s,3H),1.22(s,3H),1.16(s,3H),1.15(s,3H),0.96(s,3H),0.93(s,3H),0.76(s,3H).13C NMR(100MHz,CDCl3):δ213.68,163.45,139.42,130.71,126.20,125.42,76.66,73.38,70.16,69.45,68.14,54.73,51.42,49.72,49.46,43.50,40.13,38.09,37.78,36.50,35.74,33.09,32.70,32.64,31.95,31.29,27.22,26.30,25.00,24.28,20.32,19.54,16.46,16.33,15.78.
实施例14
化合物18的制备
将20(S)-原人参三醇化合物17(476mg)溶于吡啶中,加入0.5mL醋酐,4-二甲氨基吡啶12mg,室温下反应9小时。反应结束后加入水稀释,以乙酸乙酯萃取。有机层减压蒸干得粗产品。硅胶柱层析(石油醚/乙酸乙酯=8:1),得化合物18。1H NMR(400MHz,CDCl3):δ5.30(m,1H),5.05(t,J=6.7Hz,1H),4.88(m,1H),4.42(dd,J=11.6Hz,4.5Hz,1H),2.44(m,1H),2.00(s,6H),1.80(s,3H),1.52(s,3H),1.16(s,6H),1.12(s,3H),0.99(s,3H),0.96(s,3H),0.91(s,3H),0.87(s,3H).13C NMR(100MHz,CDCl3):δ170.89,170.48,170.22,137.42,124.55,80.09,73.88,70.84,70.38,58.62,50.83,49.70,49.27,46.51,43.69,42.44,40.88,39.39,38.30,37.77,31.97,30.35,29.30,29.22,28.43,28.18,23.19,23.05,22.02,21.29,21.14,17.20,16.87,16.83,16.72,12.23。ESI-MS:m/z 625.56[M+Na]+.
实施例15
化合物19的制备
将200mg化合物18溶于20mL二氯甲烷/甲醇1:1的混合溶剂中,通入氮气20-30min。在-78℃下通入臭氧反应15分钟至溶液变蓝即停止反应。撤去臭氧,通入氮气排出过量的O3,加入二甲硫醚淬灭反应,并将反应体系逐渐升至室温后,并在室温搅拌1小时。减压蒸干溶剂,得粗产品。硅胶柱层析(石油醚/乙酸乙酯=4:1),得化合物19。1H NMR(400MHz,CDCl3):δ5.32(m,1H),4.74(m,1H),4.43(dd,J=11.6,3.1Hz,1H),2.84(m,1H),2.28(t,J=10.9Hz,1H),2.09(s,3H),2.02(s,6H),1.89(s,3H),1.15(s,3H),1.00(s,3H),0.99(s,3H),0.93(s,3H),0.89(s,3H).13C NMR(100MHz,CDCl3):δ211.45,170.95,170.86,170.21,80.07,74.26,70.26,58.73,51.91,51.42,49.79,48.55,42.67,40.88,39.44,38.38,37.83,32.27,30.38,29.51,27.83,27.36,23.21,22.04,21.34,21.11,17.37,16.93,16.86,16.81.ESI-MS:m/z 541.41[M+Na]+.
实施例16
化合物10的制备
取100mg化合物19,加入48mg NaOH,溶于14mL二氯甲烷/甲醇/水(3:3:1)混合溶剂中。在室温下反应4小时。反应结束后加水稀释,乙酸乙酯萃取。有机层减压蒸干溶剂,粗产品柱层析(二氯甲烷/甲醇=60/1)得化合物10。13C NMR(100MHz,CD3OD):δ216.90,79.50,72.42,68.81,62.16,54.37,53.51,52.37,51.42,47.66,42.14,40.52,40.21(2C),33.54,32.83,31.42,30.39,28.48,27.78,17.85,17.53,17.15,16.10。ESI-MS:m/z 391.41[M-H]-.
实施例17
制剂的制备
片剂的制备
化合物3 0.5g;微晶纤维素10g;泊洛沙姆10g;95%乙醇50ml;羟丙基纤维素50g;25%淀粉浆100ml;硬酸镁2g,制粒,60℃干燥,12目筛整粒,压成片剂.
胶囊剂的制备
化合物3 0.5g;微晶纤维素10g;泊洛沙姆10g;95%乙醇50ml;羟丙基纤维素50g;25%淀粉浆100ml;100目筛制粒,80℃干燥,100目筛整粒,填入空胶囊。
冻干粉针剂的制备
化合物3 0.5g;泊洛沙姆5g;加入5.0g精氨酸,加入20.0g甘露醇,加入注射用水,加热溶解,稀释至1000ml,过滤、滤液超滤,分装、冷冻干燥,完毕后压盖。上述冷冻干燥分为四个阶段:(1)预冻5小时,温度在-30℃;(2)减压干燥12小时,温度在-30℃;(3)升温干燥6小时,温度在-10℃;(4)二次升温干燥4小时,温度在35℃。
达玛烷型三萜化合物及制备和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












动态评分
0.0