








专利摘要
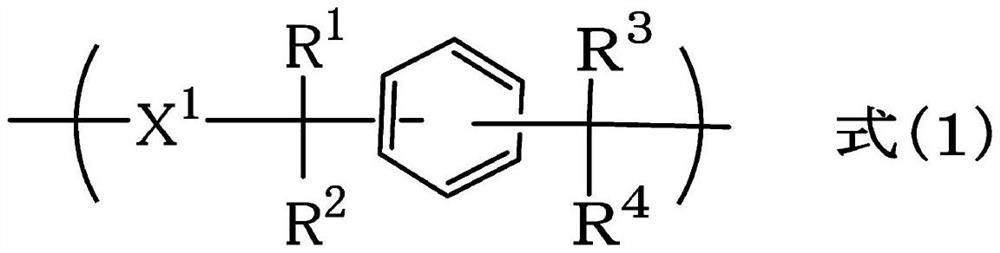
本发明的课题是提供新的抗蚀剂下层膜形成用组合物。作为解决本发明课题的方法为一种抗蚀剂下层膜形成用组合物,其包含下述聚合物和溶剂,所述聚合物具有下述式(1a)、式(1b)和式(1c)所示的重复结构单元中的任1种或2种以上,[式中,2个R1各自独立地表示烷基、烯基、芳香族烃基、卤原子、硝基或氨基,2个R2各自独立地表示氢原子、烷基、烯基、缩醛基、酰基或缩水甘油基,R3表示可以具有取代基的芳香族烃基,R4表示氢原子、苯基或萘基,在式(1b)中2个R3所表示的基团和2个R4所表示的原子或基团可以彼此不同,2个k各自独立地表示0或1,m表示3~500的整数,n、n1和n2表示2~500的整数,p表示3~500的整数,X表示单键或杂原子,2个Q各自独立地表示结构单元。]。
权利要求
1.一种抗蚀剂下层膜形成用组合物,其包含下述聚合物和溶剂,所述聚合物具有下述式(1a)、式(1b)和式(1c)所示的重复结构单元中的任1种或2种以上,
式中,2个R1各自独立地表示碳原子数1~10的烷基、碳原子数2~6的烯基、芳香族烃基、卤原子、硝基或氨基,2个R2各自独立地表示氢原子、碳原子数1~10的烷基、碳原子数2~6的烯基、缩醛基、酰基或缩水甘油基,R3表示可以具有取代基的芳香族烃基,R4表示氢原子、苯基或萘基,与同一碳原子结合的R3和R4各自表示苯基时可以彼此结合而形成芴环,在式(1b)中2个R3所表示的基团和2个R4所表示的原子或基团可以彼此不同,2个k各自独立地表示0或1,m表示3~500的整数,n、n1和n2表示2~500的整数,p表示3~500的整数,X表示单键或杂原子,2个Q各自独立地表示下述式(2)所示的结构单元,
式中,2个R1、2个R2、2个R3、2个R4、2个k、n1、n2和X与式(1b)中的含义相同,2个Q1各自独立地表示上述式(2)所示的结构单元。
2.根据权利要求1所述的抗蚀剂下层膜形成用组合物,所述聚合物为通过使至少1种的联苯酚化合物或二苯酚化合物与至少1种的芳香族醛或芳香族酮在酸催化剂的存在下进行聚合反应而合成的。
3.根据权利要求1所述的抗蚀剂下层膜形成用组合物,所述R3所表示的芳香族烃基为苯基、萘基、蒽基或芘基。
4.根据权利要求1~3的任一项所述的抗蚀剂下层膜形成用组合物,其进一步含有交联剂、酸性化合物、热产酸剂和表面活性剂中的至少1种。
5.一种半导体装置的制造方法,其包括下述工序:通过将权利要求1~4的任一项所述的抗蚀剂下层膜形成用组合物涂布于具有高低差、凹部和/或凸部的表面,进行烘烤而形成第1抗蚀剂下层膜的工序;在所述第1抗蚀剂下层膜上形成有机聚硅氧烷膜作为第2抗蚀剂下层膜的工序;在所述第2抗蚀剂下层膜上形成抗蚀剂图案的工序;将所述抗蚀剂图案作为掩模对所述第2抗蚀剂下层膜进行蚀刻的工序;将蚀刻后的所述第2抗蚀剂下层膜的图案作为掩模对所述第1抗蚀剂下层膜进行蚀刻的工序;以及将蚀刻后的所述第1抗蚀剂下层膜的图案作为掩模对所述具有高低差、凹部和/或凸部的表面进行蚀刻的工序。
说明书
技术领域
本发明涉及光刻工艺用抗蚀剂下层膜形成用组合物。特别是涉及用于形成作为掩模材使用时具有耐蚀刻性,并且具有具有高低差、凹部和/或凸部的表面的埋入性的抗蚀剂下层膜的组合物。
背景技术
在半导体装置的制造中,利用光刻工艺进行微细加工。该光刻工艺已知存在下述问题:将基板上的抗蚀剂层利用KrF准分子激光、ArF准分子激光等紫外线激光进行曝光时,由于在基板表面该紫外线激光发生反射而产生的驻波所带来的影响,不能形成具有所期望的形状的抗蚀剂图案。为了解决该问题,采用了在基板与抗蚀剂层之间设置抗蚀剂下层膜(防反射膜)。而且,作为用于形成抗蚀剂下层膜的组合物,已知使用酚醛清漆树脂。例如,专利文献1和专利文献2中公开了,含有具备将具有二苯酚基的化合物进行了酚醛清漆化的重复单元的树脂的光致抗蚀剂下层膜形成用材料。进一步,专利文献3中公开了,包含在聚合物的主链中具有3个或3个以上缩合了的芳香族环的聚合物的、能够旋转涂布的防反射膜组合物。然而,专利文献1~3中,没有记载也没有暗示包含侧链具有上述缩合了的芳香族环的聚合物的抗蚀剂下层膜形成用组合物。
为了伴随抗蚀剂图案的微细化而要求的抗蚀剂层的薄膜化,还已知形成至少2层抗蚀剂下层膜,使用该抗蚀剂下层膜作为掩模材的光刻工艺。作为形成上述至少2层的材料,可举出有机树脂(例如,丙烯酸系树脂、酚醛清漆树脂)、硅树脂(例如,有机聚硅氧烷)、无机硅化合物(例如,SiON、SiO2)。将由上述有机树脂层形成的图案作为掩模进行干蚀刻时,需要该图案相对于蚀刻气体(例如碳氟化合物)具有耐蚀刻性。作为用于形成这样的有机树脂层的组合物,例如专利文献4中公开了含有包含杂环芳香族部分的聚合物的组合物。
现有技术文献
专利文献
专利文献1:日本特开2006-259249号公报
专利文献2:日本特开2007-316282号公报
专利文献3:日本特表2010-528334号公报
专利文献4:日本特开2007-017976号公报
发明内容
发明所要解决的课题
本发明的目的在于提供用于形成对于碳氟化合物那样的蚀刻气体具有耐蚀刻性,并且具有具有高低差、凹部和/或凸部的表面的埋入性的抗蚀剂下层膜的组合物。
用于解决课题的方法
本发明的第1方式为一种抗蚀剂下层膜形成用组合物,其包含下述聚合物和溶剂,所述聚合物具有下述式(1a)、式(1b)和式(1c)所示的重复结构单元中的任1种或2种以上,
[式中,2个R1各自独立地表示碳原子数1~10的烷基、碳原子数2~6的烯基、芳香族烃基、卤原子、硝基或氨基,2个R2各自独立地表示氢原子、碳原子数1~10的烷基、碳原子数2~6的烯基、缩醛基、酰基或缩水甘油基,R3表示可以具有取代基的芳香族烃基,R4表示氢原子、苯基或萘基,与同一碳原子结合的R3和R4各自表示苯基时可以彼此结合而形成芴环,在式(1b)中2个R3所表示的基团和2个R4所表示的原子或基团可以彼此不同,2个k各自独立地表示0或1,m表示3~500的整数,n、n1和n2表示2~500的整数,p表示3~500的整数,X表示单键或杂原子,2个Q各自独立地表示下述式(2)所示的结构单元,
(式中,2个R1、2个R2、2个R3、2个R4、2个k、n1、n2和X与式(1b)中的含义相同,2个Q1各自独立地表示上述式(2)所示的结构单元。)]。
在上述X表示单键时,成为式(1a)、式(1b)和式(1c)的2个苯环通过单键而直接连接成的联苯结构。在上述X表示杂原子时,该杂原子为除了碳原子和氢原子以外的原子,为能够形成二价基团的原子,可举出例如硫原子、氧原子。在上述R1表示卤原子时,作为该卤原子,可举出例如氯原子、溴原子。上述芳香族烃基的芳香环可以为单环、多环(包含二环)、杂环中的任一者,可举出例如苯基、联苯基、萘基、蒽基、芘基、噻吩基、吡啶基。作为上述R3所表示的可以具有取代基的芳香族烃基,从耐蚀刻性和埋入性的观点出发,优选为苯基、萘基、蒽基和芘基。而且,作为芳香族烃基的取代基,可举出例如甲氧基、醛基。
本发明的抗蚀剂下层膜形成用组合物中,进一步作为任意成分,可以含有交联剂、酸性化合物、热产酸剂和表面活性剂中的至少1种。
本发明的第2方式为一种半导体装置的制造方法,其包括下述工序:通过将本发明的抗蚀剂下层膜形成用组合物涂布于具有高低差、凹部和/或凸部的表面,进行烘烤而形成第1抗蚀剂下层膜的工序;在上述第1抗蚀剂下层膜上形成有机聚硅氧烷膜作为第2抗蚀剂下层膜的工序;在上述第2抗蚀剂下层膜上形成抗蚀剂图案的工序;将上述抗蚀剂图案作为掩模对上述第2抗蚀剂下层膜进行蚀刻的工序;将蚀刻后的上述第2抗蚀剂下层膜的图案作为掩模对上述第1抗蚀剂下层膜进行蚀刻的工序;以及将蚀刻后的上述第1抗蚀剂下层膜的图案作为掩模对上述具有高低差、凹部和/或凸部的表面进行蚀刻的工序。
发明的效果
使用本发明的抗蚀剂下层膜形成用组合物而形成的抗蚀剂下层膜对于碳氟化合物那样的蚀刻气体具有耐蚀刻性,并且具有凹部和/或凸部的表面的埋入性优异。
附图说明
图1为显示使用由实施例1调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图2为显示使用由实施例2调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图3为显示使用由实施例3调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图4为显示使用由实施例4调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图5为显示使用由实施例5调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图6为显示使用由实施例6调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图7为显示使用由实施例7调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图8为显示使用由实施例8调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图9为显示使用由实施例9调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图10为显示使用由实施例10调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图11为显示使用由实施例11调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图12为显示使用由实施例12调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图13为显示使用由实施例13调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图14为显示使用由实施例14调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图15为显示使用由实施例15调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图16为显示使用由实施例16调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图17为显示使用由实施例17调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图18为显示使用由实施例18调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图19为显示使用由实施例19调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图20为显示使用由实施例20调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
图21为显示使用由比较例3调制的抗蚀剂下层膜形成用组合物来进行埋入试验的结果的截面SEM照片。
具体实施方式
作为本发明的抗蚀剂下层膜形成用组合物中所包含的具有上述式(1a)所示的重复结构单元的聚合物的结构单元,可举出例如下述式(1-1)~式(1-36)所示的结构单元,作为具有上述式(1b)所示的重复结构单元的聚合物的结构单元,可举出例如下述式(1-37)~式(1-72)所示的结构单元,作为具有上述式(1c)所示的重复结构单元的聚合物的结构单元,可举出例如下述式(1-73)~式(1-108)所示的结构单元。另外,在式(1-37)~式(1-72)中,2个Q、n1和n2与上述式(1b)中的含义相同。
本发明的抗蚀剂下层膜形成用组合物中所包含的聚合物的重均分子量以标准聚苯乙烯换算值计为例如1000~5000。
上述聚合物通过使具有2个羟基苯基的联苯酚化合物或二苯酚化合物与芳香族醛或芳香族酮在磺酸化合物等酸催化剂的存在下进行聚合反应来合成。作为具有2个羟基苯基的联苯酚化合物或二苯酚化合物,可举出例如,4,4’-联苯酚、2,2’-联苯酚、2,4’-联苯酚、4,4’-硫代二苯酚、4,4’-氧代二苯酚。作为上述聚合物的合成所使用的芳香族醛,可举出例如,糠醛、吡啶甲醛、苯甲醛、萘甲醛、蒽甲醛、菲甲醛、水杨醛、苯基乙醛、3-苯基丙醛、甲苯醛、(N,N-二甲基氨基)苯甲醛、乙酰氧基苯甲醛、1-芘甲醛、茴香醛,可以优选使用除了糠醛、吡啶甲醛那样的具有芳香族杂环的醛以外的芳香族醛。此外,上述聚合物的合成所使用的芳香族酮为二芳基酮类,可举出例如,二苯基酮、苯基萘基酮、二萘基酮、苯基甲苯基酮、二甲苯基酮、9-芴酮。上述聚合物的合成所使用的联苯酚化合物或二苯酚化合物不限定于1种化合物,可以使用2种以上,芳香族醛或芳香族酮也不限定于1种化合物,可以使用2种以上。例如,作为联苯酚化合物,可以使用4,4-联苯酚,作为芳香族醛,可以使用苯甲醛和萘甲醛。
本发明的抗蚀剂下层膜形成用组合物可以进一步含有交联剂。作为上述交联剂,可以优选使用具有至少二个交联形成用取代基的交联性化合物。可举出例如,具有羟甲基、甲氧基甲基等交联形成用取代基的、三聚氰胺系化合物、取代脲系化合物和酚系化合物。具体而言,为甲氧基甲基化甘脲、甲氧基甲基化三聚氰胺等化合物,可举出例如,四甲氧基甲基甘脲、四丁氧基甲基甘脲、六甲氧基甲基三聚氰胺。进一步,作为取代脲系化合物,可举出例如,四甲氧基甲基脲、四丁氧基甲基脲。作为酚系化合物,可举出例如,四羟基甲基联苯酚、四甲氧基甲基联苯酚(TMOM-BP)、四甲氧基甲基二苯酚。
作为上述交联剂,此外,也可以使用具有至少二个环氧基的化合物。作为这样的化合物,可举出例如,三(2,3-环氧丙基)异氰脲酸酯、1,4-丁二醇二缩水甘油基醚、1,2-环氧-4-(环氧乙基)环己烷、甘油三缩水甘油基醚、二甘醇二缩水甘油基醚、2,6-二缩水甘油基苯基缩水甘油基醚、1,1,3-三[对(2,3-环氧丙氧基)苯基]丙烷、1,2-环己烷二甲酸二缩水甘油基酯、4,4’-亚甲基双(N,N-二缩水甘油基苯胺)、3,4-环氧环己基甲基-3,4-环氧环己烷甲酸酯、三羟甲基乙烷三缩水甘油基醚、双酚A-二缩水甘油基醚、(株)ダイセル制的エポリード〔注册商标〕GT-401、エポリードGT-403、エポリードGT-301、エポリードGT-302、セロキサイド〔注册商标〕2021、セロキサイド3000、三菱化学(株)制的1001、1002、1003、1004、1007、1009、1010、828、807、152、154、180S75、871、872、日本化药(株)制的EPPN201、EPPN202、EOCN-102、EOCN-103S、EOCN-104S、EOCN-1020、EOCN-1025、EOCN-1027、ナガセケムテックス(株)制的デナコール〔注册商标〕EX-252、デナコールEX-611、デナコールEX-612、デナコールEX-614、デナコールEX-622、デナコールEX-411、デナコールEX-512、デナコールEX-522、デナコールEX-421、デナコールEX-313、デナコールEX-314、デナコールEX-321、BASFジャパン(株)制的CY175、CY177、CY179、CY182、CY184、CY192、DIC(株)制的エピクロン200、エピクロン400、エピクロン7015、エピクロン835LV、エピクロン850CRP。作为上述具有至少二个环氧基的化合物,此外,也可以使用具有氨基的环氧树脂。作为这样的环氧树脂,可举出例如,YH-434、YH-434L(新日化エポキシ制造(株)(旧东都化成(株))制)。
作为上述交联剂,此外,也可以使用具有至少2个封端异氰酸酯基的化合物。作为这样的化合物,可举出例如,三井化学(株)制的タケネート〔注册商标〕B-830、タケネートB-870N、エボニックデグサ社制的VESTANAT〔注册商标〕B1358/100。
作为上述交联剂,此外,也可以使用具有至少2个乙烯基醚基的化合物。作为这样的化合物,可举出例如,双(4-(乙烯基氧基甲基)环己基甲基)戊二酸酯、三(乙二醇)二乙烯基醚、己二酸二乙烯基酯、二甘醇二乙烯基醚、1,2,4-三(4-乙烯基氧基丁基)偏苯三酸酯、1,3,5-三(4-乙烯基氧基丁基)偏苯三酸酯、双(4-(乙烯基氧基)丁基)对苯二甲酸酯、双(4-(乙烯基氧基)丁基)间苯二甲酸酯、乙二醇二乙烯基醚、1,4-丁二醇二乙烯基醚、1,4-丁二醇二乙烯基醚、四甘醇二乙烯基醚、新戊二醇二乙烯基醚、三羟甲基丙烷三乙烯基醚、三羟甲基乙烷三乙烯基醚、己二醇二乙烯基醚、1,4-环己二醇二乙烯基醚、四甘醇二乙烯基醚、季戊四醇二乙烯基醚、季戊四醇三乙烯基醚和环己烷二甲醇二乙烯基醚。
可以添加从这些各种交联剂中选择的1种,也可以组合添加2种以上。上述交联剂的含有比例是,相对于从本发明的抗蚀剂下层膜形成用组合物中除去后述溶剂后的固体成分为例如2质量%~60质量%。
本发明的抗蚀剂下层膜形成用组合物可以进一步含有酸性化合物。上述酸性化合物作为促进交联反应的催化剂起作用,可举出例如,对甲苯磺酸、三氟甲磺酸、吡啶 -对甲苯磺酸盐、水杨酸、樟脑磺酸、5-磺基水杨酸、4-氯苯磺酸、4-羟基苯磺酸、苯二磺酸、1-萘磺酸、柠檬酸、苯甲酸、羟基苯甲酸等磺酸化合物和羧酸化合物、盐酸、硫酸、硝酸、磷酸等无机酸。可以代替上述酸性化合物,或与上述酸性化合物一起,含有热产酸剂。上述热产酸剂也作为促进交联反应的催化剂起作用,可举出例如三氟甲磺酸的季铵盐。可以添加从这些酸性化合物和热产酸剂中选择的1种,也可以组合添加2种以上。上述酸性化合物或热产酸剂的含有比例是,相对于从本发明的抗蚀剂下层膜形成用组合物中除去后述溶剂后的固体成分为例如0.1质量%~20质量%。
本发明的抗蚀剂下层膜形成用组合物可以进一步含有表面活性剂。作为上述表面活性剂,可举出例如,聚氧乙烯月桂基醚、聚氧乙烯硬脂基醚、聚氧乙烯鲸蜡基醚、聚氧乙烯油基醚等聚氧乙烯烷基醚类、聚氧乙烯辛基苯基醚、聚氧乙烯壬基苯基醚等聚氧乙烯烷基芳基醚类、聚氧乙烯/聚氧丙烯嵌段共聚物类、失水山梨糖醇单月桂酸酯、失水山梨糖醇单棕榈酸酯、失水山梨糖醇单硬脂酸酯、失水山梨糖醇单油酸酯、失水山梨糖醇三油酸酯、失水山梨糖醇三硬脂酸酯等失水山梨糖醇脂肪酸酯类、聚氧乙烯失水山梨糖醇单月桂酸酯、聚氧乙烯失水山梨糖醇单棕榈酸酯、聚氧乙烯失水山梨糖醇单硬脂酸酯、聚氧乙烯失水山梨糖醇三油酸酯、聚氧乙烯失水山梨糖醇三硬脂酸酯等聚氧乙烯失水山梨糖醇脂肪酸酯类等非离子系表面活性剂、エフトップ〔注册商标〕EF301、エフトップEF303、エフトップEF352(三菱マテリアル电子化成(株)制)、メガファック〔注册商标〕F171、メガファックF173、メガファックR-30、メガファックR-30-N(DIC(株)制)、フロラードFC430、フロラードFC431(住友スリーエム(株)制)、アサヒガード〔注册商标〕AG710、サーフロン〔注册商标〕S-382、サーフロンSC101、サーフロンSC102、サーフロンSC103、サーフロンSC104、サーフロンSC105、サーフロンSC106(旭硝子(株)制)等氟系表面活性剂、有机硅氧烷聚合物KP341(信越化学工业(株)制)。可以添加从这些表面活性剂中选择的1种,也可以组合添加2种以上。上述表面活性剂的含有比例是,相对于从本发明的抗蚀剂下层膜形成用组合物中除去后述溶剂后的固体成分为例如0.01质量%~5质量%。
本发明的抗蚀剂下层膜形成用组合物可以通过使上述各成分溶解于适当的溶剂来进行调制,以均匀的溶液状态使用。作为这样的溶剂,可举出例如,乙二醇单甲基醚、乙二醇单乙基醚、二甘醇单甲基醚、二甘醇单乙基醚、丙二醇、丙二醇单甲基醚、丙二醇单丙基醚、丙二醇单甲基醚乙酸酯、丙二醇丙基醚乙酸酯、甲基溶纤剂乙酸酯、乙基溶纤剂乙酸酯、甲苯、二甲苯、甲基乙基酮、环戊酮、环己酮、2-羟基丙酸乙酯、2-羟基-2-甲基丙酸乙酯、乙氧基乙酸乙酯、羟基乙酸乙酯、2-羟基-3-甲基丁酸甲酯、3-甲氧基丙酸甲酯、3-甲氧基丙酸乙酯、3-乙氧基丙酸乙酯、3-乙氧基丙酸甲酯、丙酮酸甲酯、丙酮酸乙酯、乙酸乙酯、乙酸丁酯、乳酸乙酯、乳酸丁酯、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮。这些有机溶剂可以使用1种,或2种以上组合使用。从上述组合物中除去有机溶剂后的固体成分的比例为例如0.5质量%~30质量%,优选为0.8质量%~15质量%。
将本发明的抗蚀剂下层膜形成用组合物涂布于具有高低差、凹部和/或凸部的表面,进行烘烤的工序如下进行:在具有具有高低差、凹部和/或凸部的表面的基板(例如,形成有布线、其它结构体的硅晶片,该硅晶片可以被氧化硅膜、氮化硅膜、氧化氮化硅膜、或铝、钨等的金属膜被覆。)上,通过旋涂器、涂布机等适当的涂布方法来涂布该组合物,然后,使用电热板等加热手段进行烘烤。作为烘烤条件,从烘烤温度100℃~400℃、烘烤时间0.3分钟~10分钟之中进行适当选择。
在通过上述工序形成的第1抗蚀剂下层膜上形成有机聚硅氧烷膜作为第2抗蚀剂下层膜,在所述第2抗蚀剂下层膜上形成抗蚀剂图案。在形成该抗蚀剂图案的工序中,曝光通过用于形成规定图案的掩模(中间掩模)来进行或利用直接描绘来进行。曝光源可以使用例如,g射线、i射线、KrF准分子激光、ArF准分子激光、EUV、电子射线。曝光后,根据需要进行曝光后加热(Post ExposureBake)。然后,通过显影液(例如2.38质量%氢氧化四甲铵水溶液)进行显影,进一步用冲洗液或纯水进行洗涤,除去所使用的显影液。然后,进行抗蚀剂图案的干燥和用于提高与基底的密合性的后烘烤。
上述抗蚀剂图案形成后进行的蚀刻工序通过干蚀刻来进行。作为干蚀刻所使用的蚀刻气体,对于第2抗蚀剂下层膜(有机聚硅氧烷膜),可举出例如CHF3、CF4、C2F6,对于由本发明的抗蚀剂下层膜形成用组合物形成的第1抗蚀剂下层膜,可举出例如O2、N2O、NO2,对于具有高低差或凹部和/或凸部的表面,可举出例如CHF3、CF4、C2F6。进一步,可以在这些气体中混合氩气、氮气或二氧化碳来使用。
以下,对于本发明,举出合成例和实施例来进行说明,但本发明不受下述记载的限定。
实施例
下述合成例1~合成例3、比较合成例1和比较合成例2所示的重均分子量和多分散度基于由凝胶渗透色谱(以下,本说明书中简称为GPC。)得到的测定结果。测定中,使用东ソー(株)制GPC装置,测定条件如下所述。
GPC柱:TSKgel SuperMultipore〔注册商标〕Hz-N(东ソー(株))
柱温度:40℃
溶剂:四氢呋喃(THF)
流量:0.35ml/分钟
标准试样:聚苯乙烯(东ソー(株))
(合成例1)
在氮气下,在300mL四口烧瓶中添加4,4’-联苯酚(15.00g,0.0806mol,东京化成工业(株)制)、1-芘甲醛(18.548g,0.0806mol,アルドリッチ社制)和对甲苯磺酸一水合物(33.2096g,0.0161mol,东京化成工业(株)制),进一步加入丙二醇单甲基醚(44.93g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(1800g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时,获得了目标的聚合物(以下,本说明书中简称为4,4’-BPOH-Py。)19.08g。4,4’-BPOH-Py的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为2200,多分散度Mw/Mn为2.10。由本合成例得到的聚合物具有式(1-4)所示的结构单元和式(1-76)所示的结构单元。
(合成例2)
在氮气下,在1000mL四口烧瓶中添加2,2’-联苯酚(70.00g,0.3759mol,东京化成工业(株)制)、1-芘甲醛(86.559g,0.3759mol,アルドリッチ社制)和甲磺酸(10.8389g,0.1126mol,东京化成工业(株)制),进一步加入丙二醇单甲基醚(167.40g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(3000g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时,获得了目标的聚合物(以下,本说明书中简称为2,2’-BPOH-Py。)84.1g。2,2’-BPOH-Py的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为1400,多分散度Mw/Mn为1.49。由本合成例得到的聚合物具有式(1-16)所示的结构单元和式(1-88)所示的结构单元。
(合成例3)
在氮气下,在200mL四口烧瓶中添加4,4’-硫代二苯酚(12.50g,0.0573mol,东京化成工业(株)制)、1-芘甲醛(13.19g,0.0573mol,东京化成工业(株)制)和甲磺酸(1.1008g,0.0115mol,关东化学(株)制),进一步加入丙二醇单甲基醚(32.74g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(1000g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时并进一步在120℃干燥24小时,获得了目标的聚合物(以下,本说明书中简称为TDP-Py。)11.6g。TDP-Py的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为1010,多分散度Mw/Mn为1.67。由本合成例得到的聚合物具有式(1-28)所示的结构单元和式(1-100)所示的结构单元。
(合成例4)
在氮气下,在300mL四口烧瓶中添加4,4’-联苯酚(10.00g,0.0537mol,东京化成工业(株)制)、1-芘甲醛(24.731g,0.1074mol,アルドリッチ社制)和对甲苯磺酸一水合物(4.2795g,0.0215mol,东京化成工业(株)制),进一步加入丙二醇单甲基醚(47.68g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(1800g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时,获得了目标的聚合物(以下,本说明书中简称为4,4’-BPOH-Py2。)21.58g。4,4’-BPOH-Py2的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为2180,多分散度Mw/Mn为2.11。由本合成例得到的聚合物具有式(1-40)所示的结构单元。
(合成例5)
在氮气下,在1000mL四口烧瓶中添加2,2’-联苯酚(25.00g,0.1343mol,东京化成工业(株)制)、1-芘甲醛(61.828g,0.2685mol,アルドリッチ社制)和甲磺酸(7.7421g,0.0806mol,东京化成工业(株)制),进一步加入丙二醇单甲基醚(94.57g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(3000g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时,获得了目标的聚合物(以下,本说明书中简称为2,2’-BPOH-Py2。)68.22g。2,2’-BPOH-Py2的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为1270,多分散度Mw/Mn为1.77。由本合成例得到的聚合物具有式(1-52)所示的结构单元。
(合成例6)
在氮气下,在200mL四口烧瓶中添加4,4’-硫代二苯酚(8.00g,0.0367mol,东京化成工业(株)制)、1-芘甲醛(16.88g,0.0773mol,东京化成工业(株)制)和甲磺酸(1.4090g,0.0147mol,关东化学(株)制),进一步加入丙二醇单甲基醚(32.13g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(1000g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时并进一步在120℃干燥24小时,获得了目标的聚合物(以下,本说明书中简称为TDP-Py2。)13.2g。TDP-Py2的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为1100,多分散度Mw/Mn为1.64。由本合成例得到的聚合物具有式(1-64)所示的结构单元。
(合成例7)
在氮气下,在500mL四口烧瓶中添加4,4’-联苯酚(30.00g,0.1611mol,东京化成工业(株)制)、1-萘甲醛(50.324g,0.3222mol,アルドリッチ社制)和对甲苯磺酸一水合物(12.8384g,0.0644mol,东京化成工业(株)制),进一步加入丙二醇单甲基醚(113.87g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇/纯水=80/20(3000g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时,获得了目标的聚合物(以下,本说明书中简称为4,4’-BPOH-Np2。)21.08g。4,4’-BPOH-Np2的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为2450,多分散度Mw/Mn为2.03。由本合成例得到的聚合物具有式(1-38)所示的结构单元。
(合成例8)
在氮气下,在100mL四口烧瓶中添加2,2’-联苯酚(7.50g,0.0403mol,东京化成工业(株)制)、1-萘甲醛(12.581g,0.0806mol,アルドリッチ社制)和甲磺酸(1.5484g,0.0161mol,东京化成工业(株)制),进一步加入丙二醇单甲基醚(26.43g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(800g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时,获得了目标的聚合物(以下,本说明书中简称为2,2’-BPOH-Np2。)5.87g。2,2’-BPOH-Np2的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为1690,多分散度Mw/Mn为2.27。由本合成例得到的聚合物具有式(1-50)所示的结构单元。
(合成例9)
在氮气下,在200mL四口烧瓶中添加4,4’-硫代二苯酚(10.00g,0.0458mol,东京化成工业(株)制)、1-萘甲醛(14.311g,0.0916mol,东京化成工业(株)制)和甲磺酸(1.7613g,0.0183mol,关东化学(株)制),进一步加入丙二醇单甲基醚(31.87g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(1000g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时并进一步在120℃干燥24小时,获得了目标的聚合物(以下,本说明书中简称为TDP-Np2。)6.13g。TDP-Np2的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为1090,多分散度Mw/Mn为1.49。由本合成例得到的聚合物具有式(1-62)所示的结构单元。
(合成例10)
在氮气下,在100mL四口烧瓶中添加2,2’-联苯酚(25.00g,0.1343mol,东京化成工业(株)制)、1-萘甲醛(10.484g,0.0671mol,アルドリッチ社制)、1-芘甲醛(15.457g,0.0671mol,东京化成工业(株)制)和甲磺酸(3.871g,0.0403mol,东京化成工业(株)制),进一步加入丙二醇单甲基醚(54.81g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至室温,在甲醇(2500g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在50℃干燥10小时,获得了目标的聚合物(以下,本说明书中简称为2,2’-BPOH-NpPcA。)28.02g。2,2’-BPOH-NpPcA的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为2000,多分散度Mw/Mn为2.12。推定由本合成例得到的聚合物具有式(1-14)、(1-16)、(1-86)、(1-88)所示的结构单元。
(比较合成例1)
在氮气下,在100mL四口烧瓶中添加1,1-双(4-羟基苯基)环己烷(6.00g,0.0224mol,东京化成工业(株)制)、甲醛36%溶液(1.87g,0.0224mol,东京化成工业(株)制)和甲磺酸(0.1075g,0.0011mol,东京化成工业(株)制),进一步加入丙二醇单甲基醚(6.69g,关东化学(株)制),进行搅拌,升温至120℃使其溶解,开始聚合。24小时后放冷至60℃,添加四氢呋喃(13g,关东化学(株)制)进行稀释,在甲醇(320g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在80℃干燥24小时,获得了目标的具有下述式(3)所示的结构单元的聚合物(以下,本说明书中简称为BHPCH-FA。)4.45g。BHPCH-FA的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为10700,多分散度Mw/Mn为3.64。
(比较合成例2)
在氮气下,在100mL四口烧瓶中添加咔唑(6.69g,0.040mol,东京化成工业(株)制)、9-芴酮(7.28g,0.040mol,东京化成工业(株)制)和对甲苯磺酸一水合物(0.76g,0.0040mol,东京化成工业(株)制),进一步加入1,4-二 烷(6.69g,关东化学(株)制),进行搅拌,升温至100℃使其溶解,开始聚合。24小时后放冷至60℃,添加氯仿(34g,关东化学(株)制)进行稀释,在甲醇(168g,关东化学(株)制)中使其再沉淀。将所得的沉淀物进行过滤,利用减压干燥机在80℃干燥24小时,获得了目标的具有下述式(4)所示的结构单元的聚合物(以下,本说明书中简称为PCzFL。)9.37g。PCzFL的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为2800,多分散度Mw/Mn为1.77。
(实施例1)
在由合成例1获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例2)
在由合成例1获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g、作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例3)
在由合成例2获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例4)
在由合成例2获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g、作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例5)
在由合成例3获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例6)
在由合成例3获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g和作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例7)
在由合成例4获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例8)
在由合成例4获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g、作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例9)
在由合成例5获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例10)
在由合成例5获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g、作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例11)
在由合成例6获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例12)
在由合成例6获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g、作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例13)
在由合成例7获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例14)
在由合成例7获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g、作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例15)
在由合成例8获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例16)
在由合成例8获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,成分为三氟甲磺酸的季铵盐)0.10g,作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例17)
在由合成例9获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例18)
在由合成例9获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g、作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例19)
在由合成例10获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(实施例20)
在由合成例10获得的聚合物20g中混合作为交联剂的TMOM-BP(本州化学工业(株)制,3,3’,5,5’-四甲氧基甲基-4,4’-二羟基联苯)2.0g、作为催化剂的热产酸剂TAG-2689(King社制,三氟甲磺酸的季铵盐)0.10g、作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于环己酮88g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(比较例1)
在市售的甲酚酚醛清漆树脂(使用甲酚和甲醛而获得的酚醛清漆树脂)20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于丙二醇单甲基醚乙酸酯80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。本比较例中所使用的甲酚酚醛清漆树脂的、由GPC得到的聚苯乙烯换算测定的重均分子量Mw为4000,多分散度Mw/Mn为2.1。
(比较例2)
在由比较合成例1获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于丙二醇单甲基醚乙酸酯80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(比较例3)
在由比较合成例2获得的聚合物20g中混合作为表面活性剂的メガファックR-30(DIC(株)制)0.06g,使其溶解于丙二醇单甲基醚乙酸酯80g,制成溶液。然后,使用孔径0.10μm的聚乙烯制微型过滤器进行过滤,然后,使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,调制出光刻工艺所使用的抗蚀剂下层膜形成用组合物。
(在光致抗蚀剂溶剂中的溶出试验)
使用旋转涂布机,将由实施例1~实施例20和比较例1调制的抗蚀剂下层膜形成用组合物分别涂布于硅晶片上。在电热板上在400℃烘烤2分钟,形成了抗蚀剂下层膜(膜厚0.25μm)。将该抗蚀剂下层膜浸渍于作为使用于抗蚀剂的溶剂的乳酸乙酯、丙二醇单甲基醚、丙二醇单甲基醚乙酸酯和环己酮中,确认了在这些溶剂中不溶。
(干蚀刻速度的测定)
干蚀刻速度的测定所使用的蚀刻器和蚀刻气体如下。
蚀刻器:ES401(日本サイエンティフィック(株)制)
蚀刻气体:CF4
使用旋转涂布机,将由实施例1~实施例20、比较例1和比较例2调制的抗蚀剂下层膜形成用组合物分别涂布于硅晶片上。在电热板上在240℃烘烤1分钟,或在400℃烘烤2分钟,形成了抗蚀剂下层膜(膜厚0.25μm)。使用CF4气体作为蚀刻气体,测定这些抗蚀剂下层膜的干蚀刻速度。此外,使用旋转涂布机,将苯酚酚醛清漆树脂(市售品,由GPC得到的聚苯乙烯换算测定的重均分子量Mw为2000,多分散度Mw/Mn为2.5)溶液涂布于硅晶片上,在电热板上在205℃烘烤1分钟而形成了苯酚酚醛清漆树脂膜(膜厚0.25μm)。使用CF4气体作为蚀刻气体,测定该苯酚酚醛清漆树脂膜的干蚀刻速度。以干蚀刻速度比的方式算出将该苯酚酚醛清漆树脂膜的干蚀刻速度设为1.00时的、由实施例1~实施例20、比较例1和比较例2调制的抗蚀剂下层膜形成用组合物形成的抗蚀剂下层膜的干蚀刻速度,将结果示于表1中。干蚀刻速度比越小,则表示相对于CF4气体的耐蚀刻性越高。
干蚀刻速度比=(抗蚀剂下层膜的干蚀刻速度)/(苯酚酚醛清漆树脂膜的干蚀刻速度)
[表1]干蚀刻速度比
(对空孔晶片(ホールウェハー)的埋入性试验)
使用旋转涂布机,将由实施例1~实施例20和比较例3调制的抗蚀剂下层膜形成用组合物分别涂布于空孔晶片上。在电热板上在400℃烘烤2分钟,形成了抗蚀剂下层膜(空孔图案未形成部的膜厚0.25μm)。作为上述空孔晶片,使用形成有直径100nm、高度400nm的空孔图案的晶片。如上述那样分别涂布由实施例1~实施例20调制的抗蚀剂下层膜形成用组合物,由采用扫描型电子显微镜(SEM)观察烘烤后的各空孔晶片的截面得到的图1~图20所示的截面SEM照片可知,甚至连空孔内部都被抗蚀剂下层膜充分地填充了。另一方面,如上述那样涂布由比较例3调制的抗蚀剂下层膜形成用组合物,由采用扫描型电子显微镜观察烘烤后的空孔晶片的截面得到的图21所示的截面SEM照片可知,空孔内部局部地存在有空洞。
抗蚀剂下层膜形成用组合物专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












动态评分
0.0