









专利摘要
本发明实施例提供了一种从待测样品中分离126Sn的分离方法,包括以下步骤:将氟化氢铵和待测样品按预定质量比混合;对上述步骤获得的混合物进行密闭并加热;加热结束、待冷却后向所述混合物中加入酸,使所述混合物中的有机物分解;待所述有机物分解后,向所述混合物中加入盐酸进行溶解;从所述溶解液中萃取126Sn。该分离方法适用于从放射性废物中分离126Sn。根据本发明实施例的126Sn的分离方法,分离流程简单,126Sn回收率高,有利于提高126Sn测量精度;同时,该方法也适用于环境样品中锡的分离回收。
权利要求
1.一种从待测样品中分离
将氟化氢铵和待测样品按预定质量比混合;
对上述步骤获得的混合物进行密闭并加热;
加热结束、待冷却后向所述混合物中加入酸,使所述混合物中的有机物分解;
待所述有机物分解后,向所述混合物中加入盐酸进行溶解;
从所述溶解液中萃取
2.根据权利要求1所述的分离方法,其中,
所述萃取步骤中,采用氢氟酸反萃取有机相中的
3.根据权利要求2所述的分离方法,其中,
在所述溶解步骤中,所述盐酸的浓度为4~9mol/L;
在进行所述盐酸溶解后,向溶解液中加入硫酸或硫酸盐。
4.根据权利要求1所述的分离方法,还包括:
对所述待测样品进行预处理;
所述预处理步骤包括:将所述待测样品干燥,然后研磨至预定颗粒尺寸。
5.根据权利要求1所述的分离方法,还包括:
对获得的
6.根据权利要求5所述的分离方法,其中,所述纯化步骤包括:
采用盐酸溶解萃取步骤中分离获得的产物;
对溶解液进行阴离子交换纯化。
7.根据权利要求6所述的分离方法,其中,
将所述溶解液过HZ201阴离子交换树脂柱,盐酸淋洗;
采用氢氟酸-盐酸解吸
8.根据权利要求1-7任一项所述的分离方法,所述方法适用于从放射性废物中分离
9.一种从待测样品中分离锡的方法,所述方法采用权利要求1-8任一项所述的分离方法,所述待测样品包括:岩石、土壤、污泥和水体样品。
说明书
技术领域
本发明涉及核素分离技术领域,具体涉及一种从待测样品中分离
背景技术
传统用于样品溶解的方法包括微波消解法、碱熔融法。其中,微波消解法常采用试剂有硝酸、盐酸和氢氟酸等,但SnCl4的沸点只有114℃,在蒸发过程中容易造成Sn的损失,并且微波消解法无法保证溶解完全,从而影响测量及分析的准确性;碱熔融法虽可实现全溶解,但需要高温反应条件(一般在500℃左右),容易造成放射性污染。
基于此,提出改进的样品溶解方法,以便为后续分离步骤提供有利的基础。同时,结合优化的分离、纯化工艺,实现待测样品中
发明内容
本发明的实施例提出一种从待测样品中分离
根据本发明的一个方面,提供一种从待测样品中分离
根据本发明的实施例,所述萃取步骤中,萃取剂为磷酸三异戊脂。
根据本发明的实施例,所述萃取步骤中,采用氢氟酸反萃取有机相中的
根据本发明的实施例,在所述溶解步骤中,所述盐酸的浓度为4~9mol/L;在进行所述盐酸溶解后,向溶解液中加入硫酸或硫酸盐。
根据本发明的实施例,所述分离方法还包括:对所述待测样品进行预处理;所述预处理步骤包括:将所述待测样品干燥,然后研磨至预定颗粒尺寸。
根据本发明的实施例,所述分离方法还包括:对获得的
根据本发明的实施例,所述纯化步骤包括:采用盐酸溶解萃取步骤中分离获得的产物;对溶解液进行阴离子交换纯化。
根据本发明的实施例,将所述溶解液过HZ201阴离子交换树脂柱,盐酸淋洗;采用氢氟酸-盐酸解吸
根据本发明的实施例,所述分离方法适用于从放射性废物中分离
根据本发明的另一个方面,还提供一种从待测样品中分离锡的方法,所述方法采用上述实施例所述的
基于上述技术方案,本发明实施例提供的
附图说明
通过下文中参照附图对本发明所作的描述,本发明的其它目的和优点将显而易见,并可帮助对本发明有全面的理解。
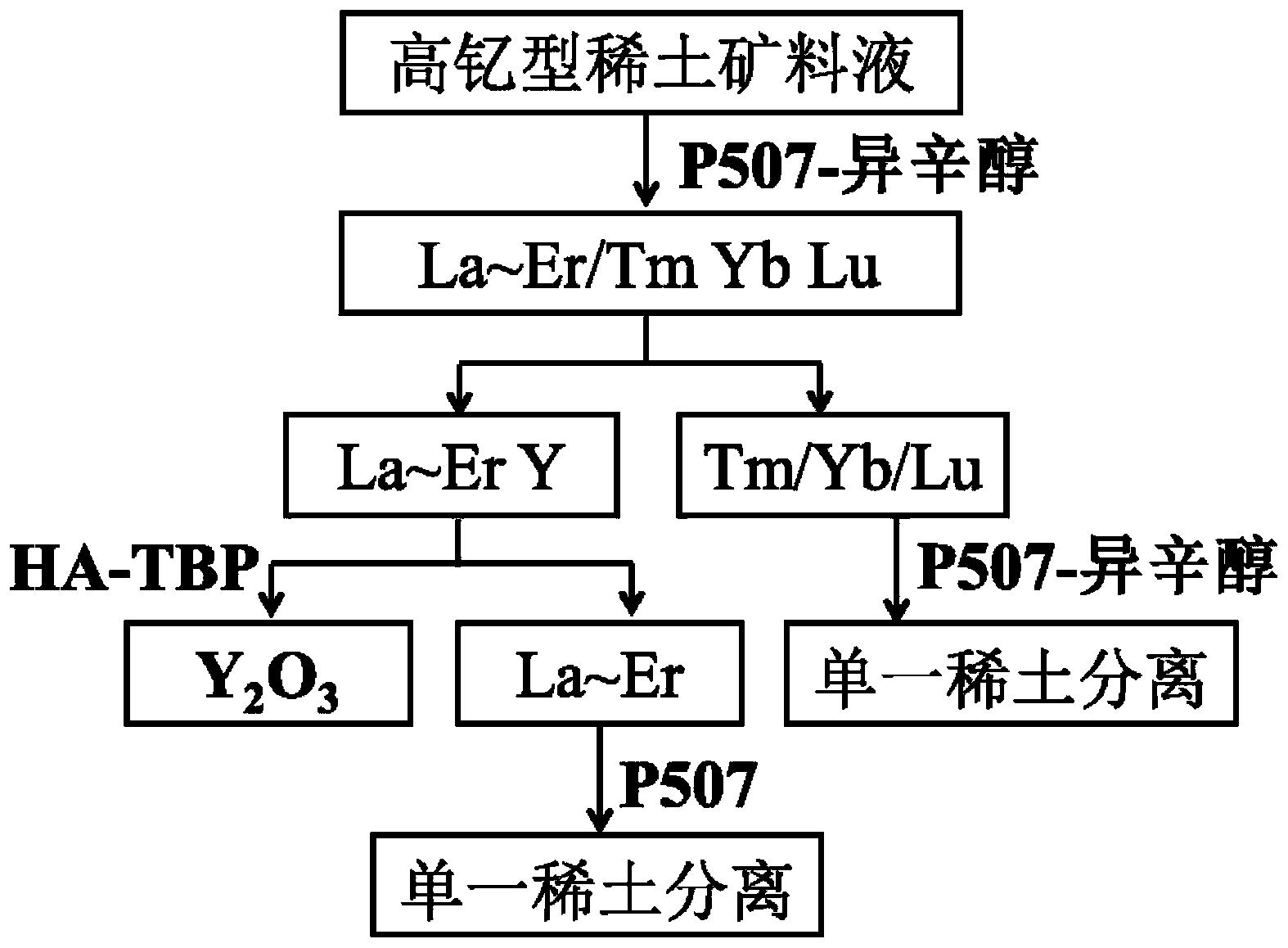
图1示意性示出了根据本发明一个实施例的从待测样品中分离
图2示意性示出了根据本发明另一个实施例的从待测样品中分离
图3示意性示出了根据本发明另一个实施例的从待测样品中分离
需要说明的是,附图并不一定按比例来绘制,而是仅以不影响读者理解的示意性方式示出。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明实施例的附图,对本发明的技术方案进行清楚、完整地描述。显然,所描述的实施例是本发明的一个实施例,而不是全部的实施例。基于所描述的本发明的实施例,本领域普通技术人员在无需创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
除非另外定义,本发明使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。
根据本发明的实施例,为了实现待测样品中
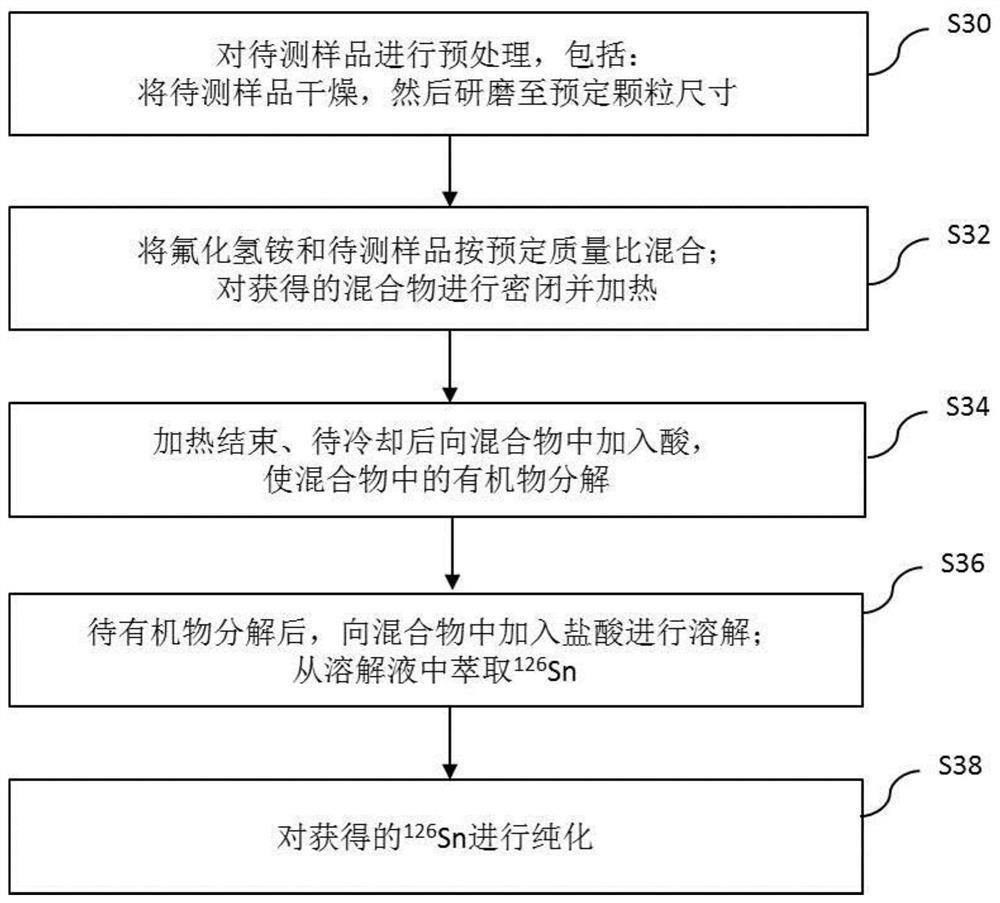
图1示意性示出了根据本发明实施例的从待测样品中分离
如图1所示,该方法包括操作S12~S16。
在操作S12,将氟化氢铵和待测样品按预定质量比混合;对获得的混合物进行密闭并加热。
在操作S14,加热结束、待冷却后向混合物中加入酸,使混合物中的有机物分解。
在操作S16,待有机物分解后,向混合物中加入盐酸进行溶解;从溶解液中萃取
根据本发明实施例,采用氟化氢铵作为主溶解剂对待测样品进行溶解,不仅可实现待测样品全溶解,该溶解方法过程中不涉及高压、在常压下可进行,可避免使用氢氟酸从而降低对人体危害,具有安全、方便操作等优点。
例如,根据待测样品质量,取适量氟化氢铵,将混合物置于消解容器中、密闭,轻微摇晃混合后,置于加热设备中进行预定时间加热。
消解结束后,待温度冷却,将消解容器取出,可向混合物中加入酸,以除去混合物中的有机物杂质。例如向混合物中加入酸,置于电热板上加热预定时间,然后蒸发至近干状态;可重复此过程1~3次,以充分除尽有机物。
在有机物除杂步骤中,例如采用一定浓度的硝酸,或者还可以在硝酸中加入少量高氯酸,以提高有机物分解效率。
进一步的,待有机物分解后,向混合物中加入盐酸进行溶解;采用盐酸介质,有利于待萃取物
根据本发明实施例,在操作S16中,萃取剂例如采用磷酸三异戊脂。
相比常用的萃取剂磷酸三丁酯(TBP),磷酸三异戊脂(TiAP)在水中的溶解度只有TBP在水中溶解度的1/19.5,即TiAP密度与水的密度相差大,作为萃取剂从水溶液中萃取目标元素时,更利于两相的分离。由此,可减少萃取过程中
进一步的,采用氢氟酸反萃取有机相中的
根据本发明实施例,在操作S16中,为提高TiAP萃取分配系数,采用的盐酸浓度例如为4~9mol/L;或者可以在进行盐酸溶解后,向溶解液中加入少量硫酸或硫酸盐,进一步提高分配系数。
图2示意性示出了根据本发明另一个实施例的从待测样品中分离
如图2所示,分离方法相比图1中分离方法,增加了操作S20:对待测样品进行预处理;预处理步骤例如包括:将待测样品干燥,然后研磨至预定颗粒尺寸。
对待测样品进行研磨,可提高其在氟化氢铵溶解步骤中的溶解效率,例如加快溶解,并使得溶解充分,为后续分离步骤提供有利的基础。
图3示意性示出了根据本发明另一个实施例的从待测样品中分离
如图3所示,分离方法相比图2中分离方法,增加了操作S38:对获得的
由于放射性污染物或废物中存在的高活度的
根据本发明实施例,纯化方法例如采用阴离子交换法。例如先采用盐酸溶解萃取步骤中分离获得的产物,然后选择阴离子交换树脂进行纯化。
根据本发明实施例,在操作S38中,纯化流程为:将S36步骤中获得的反萃液蒸干,加入盐酸进行溶解;将溶解液过HZ201阴离子交换树脂柱,并采用盐酸淋洗;然后用氢氟酸-盐酸从树脂中洗脱
其中,HZ201是一种强碱性阴离子交换树脂,其在盐酸介质下对Sn强烈吸附,采用低浓度盐酸淋洗时可将干扰高纯锗测量的干扰核素
进一步的,采用上述实施例的分离方法分离获得的
上述实施例的分离方法适用于从放射性废物中分离
本发明实施例对分离步骤中萃取剂、阴离子交换树脂的种类不作限定,只要能够实现提高
根据本发明实施例,上述用于
以下结合具体实施例对本发明技术方案进一步说明。
从待测样品中分离
S100:将待测样品烘干并研磨至200目;
S102:称取研磨好的待测样品和氟化氢铵一同置于消解罐内胆中(待测样品:氟化氢铵=1:5,质量比),将内胆密闭后,置于鼓风干燥箱中235℃下加热6h;
S104:加热结束,待温度冷却后,取出内胆,加入适量硝酸,盖上内胆盖,在电热板上220℃下加热2~3h;
冷却后,取下内胆盖,在电热板上220℃下蒸干;
S106:采用4~9mol/L盐酸对上述混合物进行溶解;
采用TiAP萃取溶解液中
采用氢氟酸反萃取有机相中的
S108:将上一步骤获得的反萃液蒸干,加入浓度大于1mol/L盐酸进行溶解;
将溶解液过HZ201阴离子交换树脂柱,采用1mol/L盐酸淋洗;然后采用3moll/L氢氟酸-2moll/L盐酸从树脂中洗脱
进一步的,将分离结束获得的解吸液进行蒸干(在低于150℃电热板上蒸干),然后用酸溶解,然后可进行高纯锗γ能谱或质谱测量。
实施例1:
采用上述分离方法对待测样品中Sn提取,验证其定量回收率。
(1)称取0.5063g已经烘干并研磨至200目的未知土壤及0.5078g、0.5110g标准石英砂岩(GBW07106)至三个消解罐内胆中;
向未知土壤中滴加0.05133g高丰度
分别向3个消解罐内胆中加入2.5g氟化氢铵,摇匀;另取一个消解罐内胆直接加入2.5g氟化氢铵;
采用上述氟化氢铵溶解法进行溶解,最后均用30mL 6mol/L盐酸溶解。
(2)将四个消解罐内胆获得的溶解液,均用15mL TiAP萃取10min,离心分离油相,再用15mL TiAP萃取10min;合并两次萃取的油相。
(3)用15mL 0.2mol/L氢氟酸反萃10min,离心分离水相,再用15mL 0.2mol/L氢氟酸反萃10min;合并两次反萃的水相。
(4)反萃液蒸干,用10mL 0.5mol/L盐酸溶解。
对上述分离产物进行质谱测量,结果见表1。
表1标准岩石GBW07106及未知土壤中Sn的分析结果
由表1可知,标准岩石GBW07106中Sn的证书给出值为1.1±0.2μg/g,即1-标准岩石中Sn的(理论)含量应为0.56±0.1μg;而由测量结果可知,直接分离出的产物中Sn含量为0.74μg,减去4-空白实测流程的空白值(0.18μg)可得实际测量的1-标准岩石中分离的Sn含量为0.56μg。这表明溶解过程中没有造成Sn损失,并且采用TiAP能够将溶解液中Sn定量提取出。
可见,采用本发明萃取流程,能够从待测样品中定量回收Sn,Sn的收率可达100%;由此采用本发明的分离方法,可实现放射性废物中
实施例2:
采用上述分离方法对待测样品中放射性Sn提取,测量其收率和干扰核素的去污因子。
(1)称取0.5029g已经烘干并研磨至200目的未知土壤至消解罐内胆中,滴加
采用上述氟化氢铵溶解法进行溶解,最后均用30mL 6mol/L盐酸溶解。
(2)将两个消解罐内胆获得的溶解液,均用15mL TiAP萃取10min,离心分离油相,再用15mL TiAP萃取10min;合并两次萃取的油相。
(3)用15mL 0.2mol/L氢氟酸反萃10min,离心分离水相,再用15mL 0.2mol/L氢氟酸反萃10min;合并两次反萃的水相。
(4)反萃液蒸干,用2mL 6mol/L盐酸溶解。
(5)将上一步骤获得的溶解液上HZ201树脂柱 20mL 1mol/L盐酸淋洗,20mL 3moll/L氢氟酸-2mol/L盐酸解吸锡。
用高纯锗测量
表2指示剂验证分离流程对Sn的收率及对Cs、Eu和Am的去污效果
由表2可知,采用
采用本发明的分离方法,可实现待测放射性污染物中
对于本发明的实施例,还需要说明的是,在不冲突的情况下,本发明的实施例及实施例中的特征可以相互组合以得到新的实施例。
以上,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,本发明的保护范围应以权利要求的保护范围为准。
从待测样品中分离Sn的分离方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0