

IPC分类号 : C09B57/14,C07D311/78,C09K11/06,G01N21/64






专利摘要
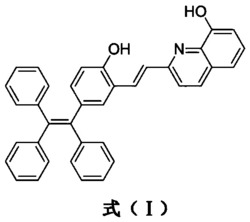
本发明公开了一种新型近红外的高量子产率染料,该染料具有如下结构式I:R1、R2、R6、R7分别独立的为氢、低级烃基、醚基、取代烷基或酰基;R3、R4、R5、R8、R9、R10分别独立的为氢、低级烃基或卤素;R11为氢、甲基、氰基、或三氟甲基;为阴离子;还公开了该染料的制备方法。该染料具有良好的生物相容性、低毒性、较长的荧光发射和很高的荧光量子产率,避免了背景自发荧光,可获得高信噪比的荧光成像图,用于生物体系中核仁RNA荧光标记,对研究与核仁RNA相关的生理学过程有着重要的作用。
权利要求
1.一种近红外的高量子产率染料,其特征在于,具有如下结构式I:
R1、R2、R6、R7分别独立的为氢、低级烃基、醚基、取代烷基或酰基;R3、R4、R5、R8、R9、R10分别独立的为氢、低级烃基或卤素;R11为氢、甲基、氰基或三氟甲基; 为阴离子;
所述R1、R2、R6、R7中低级烃基为直链、支链或环状;所述R1、R2、R6、R7中低级烃基包含1-6个碳原子;
所述醚基中碳原子数为3-6,氧原子数≤2;
所述取代烷基为直链或支链;所述取代烷基为ω-甲酸酯基取代1-6个碳原子烷基、ω-甲酰胺基取代1-6个碳原子烷基、ω-卤素取代1-6个碳原子烷基、ω-羟基取代1-6个碳原子烷基、ω-氨基取代1-6个碳原子烷基或ω-巯基取代1-6个碳原子烷基;其中,所述ω-甲酸酯基取代1-6个碳原子烷基中甲酸酯基为2-5个碳原子烷基甲酸酯基;所述的ω-甲酰胺基取代1-6个碳原子烷基中甲酰胺基为2-5个碳原子烃基甲酰胺基;
所述酰基为2-6个碳原子烷基酰基、叔丁氧羰基、苯甲酰基、1-6个碳原子取代苯甲酰基或卤素取代苯甲酰基;
所述卤素为氟、氯、溴或碘;
所述R3、R4、R5、R8、R9、R10中的低级烃基为甲基或乙基;
所述 为平衡电荷的任意有机或无机阴离子;
其中,R1与R3、R1与R10、R2与R3、R2与R10、R6与R5、R6与R8、R7与R5、R7与R8、R1与R2或R6与R7能独立形成如下Ia-In结构:
其中R为氢、甲基或乙基;Y为C、O或NR。
2.根据权利要求1所述的染料,其特征在于,
所述的R1、R2、R6、R7中的低级烃基为甲基、乙基、丙基、异丙基、环丙基、烯丙基、丁基、异丁基或叔丁基;
所述醚基为CH2CH2OCH3、CH2CH2OCH2CH3、CH2CH2OCH2CH3OH、CH2CH2OCH2CH3OCH3或CH2CH2OCH2CH3OCH2CH3;
所述ω-2-5个碳原子烷基甲酸酯基取代1-6个碳原子烷基为(CH2)mCOO(CH2)nCH3或(CH2)mCOOC(CH3)3,其中m为1-4,n为0-3;
所述ω-2-5个碳原子烃基甲酰胺基取代1-6个碳原子烷基为(CH2)mCONH(CH2)nCH3或(CH2)mCON[(CH2)nCH3]2,其中m为1-4,n为0-3;
所述ω-卤素取代1-6个碳原子烷基为(CH2)mCl、(CH2)mBr或(CH2)mI,其中m为1-6;
所述ω-羟基取代1-6个碳原子烷基为(CH2)mOH,其中m为1-6;
所述ω-氨基取代1-6个碳原子烷基为(CH2)mNH2,其中m为1-6;
所述ω-巯基取代1-6个碳原子烷基为(CH2)mSH,其中m为1-6;
所述2-6个碳原子烷基酰基为乙酰基、丙酰基、丁酰基或叔丁酰基;
所述1-6个碳原子取代苯甲酰基为甲基取代苯甲酰基、乙基取代苯甲酰基、丙基取代苯甲酰基、丁基取代苯甲酰基或叔丁基取代苯甲酰基;
所述卤素取代苯甲酰基为氯取代苯甲酰基、溴取代苯甲酰基或碘取代苯甲酰基。
3.如权利要求1所述的近红外的高量子产率染料的制备方法,其特征在于,结构式I中R11为氢或三氟甲基的染料的制备包括如下步骤:
将1毫摩尔化合物II与1毫摩尔化合物III加入到5-15毫升浓硫酸中,在加热条件下反应1-4小时,冷却后将反应液慢慢滴加20-100倍体积的冰水中,再加入0.01-1倍体积的质量百分浓度为70%的高氯酸,充分搅拌,析出固体,过滤、真空干燥,柱层析分离得产物;
所述加热的温度为60-100℃;
所述化合物II的结构式如下:
所述化合物III的结构式如下:
其中,R1、R2、R6、R7、R3、R4、R5、R8、R9、R10的定义如权利要求1;R0为氢或三氟甲基。
4.如权利要求1所述的近红外的高量子产率染料的制备方法,其特征在于,结构式I中R11为甲基的染料的制备包括如下步骤:
1)将1毫摩尔化合物II与1毫摩尔化合物III加入到5-15毫升浓硫酸中,在加热条件下反应1-4小时,冷却后将反应液慢慢滴加20-100倍体积的冰水中,再加入0.01-1倍体积的质量百分浓度为70%的高氯酸,充分搅拌,析出固体,过滤、真空干燥,柱层析分离得产物1-a;
2)将1毫摩尔I-a溶于5毫升无水THF中,在N2保护条件下逐滴加入3-6毫升中含有1毫摩尔CH3MgI的THF溶液,常温搅拌24h后,再加入2-5毫升含有1毫摩尔DDQ的THF溶液,继续搅拌1-12h,将反应液旋干后柱层析分离得到产物;
所述加热的温度为60-100℃;
所述化合物II的结构式如下:
所述化合物III的结构式如下:
其中,R1、R2、R6、R7、R3、R4、R5、R8、R9、R10的定义如权利要求1;R0为氢。
5.如权利要求1所述的近红外的高量子产率染料的制备方法,其特征在于,结构式I中R11为氰基的染料的制备包括如下步骤:
①将1毫摩尔化合物II与1毫摩尔化合物III加入到5-15毫升浓硫酸中,在加热条件下反应1-4小时,冷却后将反应液慢慢滴加20-100倍体积的冰水中,再加入0.01-1倍体积的质量百分浓度为70%的高氯酸,充分搅拌,析出固体,过滤、真空干燥,柱层析分离得产物1-a;
②将1毫摩尔得到的I-a溶于5-20毫升无水DMF中,再加入1毫摩尔的NaCN,常温搅拌24h后,再加入2-5毫升中含有1毫摩尔DDQ的THF溶液,将反应液旋干后柱层析分离得到产物;
所述加热的温度为60-100℃;
所述化合物II的结构式如下:
所述化合物III的结构式如下:
其中,R1、R2、R6、R7、R3、R4、R5、R8、R9、R10的定义如权利要求1;R0为氢。
6.根据权利要求3-5任一项所述的制备方法,其特征在于,
所述R1、R2、R6、R7中低级烃基为直链、支链或环状;所述R1、R2、R6、R7中低级烃基包含1-6个碳原子;
所述醚基中碳原子数为3-6,氧原子数≤2;
所述取代烷基为直链或支链;所述取代烷基为ω-甲酸酯基取代1-6个碳原子烷基、ω-甲酰胺基取代1-6个碳原子烷基、ω-卤素取代1-6个碳原子烷基、ω-羟基取代1-6个碳原子烷基、ω-氨基取代1-6个碳原子烷基或ω-巯基取代1-6个碳原子烷基;其中,所述ω-甲酸酯基取代1-6个碳原子烷基中甲酸酯基为2-5个碳原子烷基甲酸酯基;所述的ω-甲酰胺基取代1-6个碳原子烷基中甲酰胺基为2-5个碳原子烃基甲酰胺基;
所述酰基为2-6个碳原子烷基酰基、叔丁氧羰基、苯甲酰基、1-6个碳原子取代苯甲酰基或卤素取代苯甲酰基;
所述卤素为氟、氯、溴或碘;
所述R3、R4、R5、R8、R9、R10中的低级烃基为甲基或乙基。
7.根据权利要求3所述的制备方法,其特征在于,所述化合物II和化合物III能独立形成如下IIa-IIg和IIIa-IIIg结构:
其中R为氢、甲基或乙基;Y为C、O或NR;
R1、R2、R6、R7、R3、R4、R5、R8、R9、R10的定义如权利要求1;R0为氢或三氟甲基。
8.根据权利要求4所述的制备方法,其特征在于,所述化合物II和化合物III能独立形成如下IIa-IIg和IIIa-IIIg结构:
其中R为氢、甲基或乙基;Y为C、O或NR;
R1、R2、R6、R7、R3、R4、R5、R8、R9、R10的定义如权利要求1;R0为氢。
9.根据权利要求5所述的制备方法,其特征在于,所述化合物II和化合物III能独立形成如下IIa-IIg和IIIa-IIIg结构:
其中R为氢、甲基或乙基;Y为C、O或NR;
R1、R2、R6、R7、R3、R4、R5、R8、R9、R10的定义如权利要求1;R0为氢。
10.根据权利要求3-5任一项所述的制备方法,其特征在于,
所述R1、R2、R6、R7中的低级烃基为甲基、乙基、丙基、异丙基、环丙基、烯丙基、丁基、异丁基或叔丁基;
所述醚基为CH2CH2OCH3、CH2CH2OCH2CH3、CH2CH2OCH2CH3OH、CH2CH2OCH2CH3OCH3或CH2CH2OCH2CH3OCH2CH3;
所述ω-2-5个碳原子烷基甲酸酯基取代1-6个碳原子烷基为(CH2)mCOO(CH2)nCH3或(CH2)mCOOC(CH3)3,其中m为1-4,n为0-3;
所述ω-2-5个碳原子烃基甲酰胺基取代1-6个碳原子烷基为(CH2)mCONH(CH2)nCH3或(CH2)mCON[(CH2)nCH3]2,其中m为1-4,n为0-3;
所述ω-卤素取代1-6个碳原子烷基为(CH2)mCl、(CH2)mBr或(CH2)mI,其中m为1-6;
所述ω-羟基取代1-6个碳原子烷基为(CH2)mOH,其中m为1-6;
所述ω-氨基取代1-6个碳原子烷基为(CH2)mNH2,其中m为1-6;
所述ω-巯基取代1-6个碳原子烷基为(CH2)mSH,其中m为1-6;
所述2-6个碳原子烷基酰基为乙酰基、丙酰基、丁酰基或叔丁酰基;
所述1-6个碳原子取代苯甲酰基为甲基取代苯甲酰基、乙基取代苯甲酰基、丙基取代苯甲酰基、丁基取代苯甲酰基或叔丁基取代苯甲酰基;
所述卤素取代苯甲酰基为氯取代苯甲酰基、溴取代苯甲酰基或碘取代苯甲酰基。
11.如权利要求1-2任一项所述的近红外的高量子产率染料在制备近红外的核仁RNA荧光标记探针中的应用。
说明书
技术领域
本发明涉及近红外的核仁RNA染料,提供了一种新型近红外的高量子产率的染料、制备及应用。
背景技术
由于其高时空分辨率以及快速和非侵入式操作,小分子荧光染料基于的荧光成像已经成为强大的手段对生命系统中生物现象(比如生物分子检测,标记,癌症诊断和治疗等)的可视化观察。RNA是生命系统中特别重要的生物分子,并且细胞核中的RNA位于核仁中,它们在基因编码,转录和表达起着至关重要的作用。生物化学过程中的许多生理过程包括细胞增殖和分化等都与RNA的功能紧密相关。因此,对RNA实时的成像在生化研究中具有重要的意义。许多荧光成像技术(RNA微注射,荧光原位杂交(FISH)或是绿色荧光蛋白(GFP)及其衍生物黄色荧光蛋白(YFP)标记)已经利用来实现活细胞内RNA可视化,但是这些技术具有差的细胞通透性并且可能影响细胞的正常功能。因此,开发出具有好的光物理性质、高的细胞穿透性和易于操作的小分子染料对RNA选择性成像具有重要意义。
目前为止,Invitrogen公司的具有绿色荧光发射的RNASelect是唯一用于细胞成像的商用RNA探针,但是其应用不是很广泛,染色选择性较差,荧光波长也比较短,该探针的分子结构及合成方法至今尚未公开。近年来研究人员通过多种努力开发了很多RNA标记的染料。大多数情况下,在活细胞中获得一张满意的RNA成像需要较长的孵育时间(几十分钟甚至几个小时),只有几种具有正电荷的染料能够顺利的进入细胞并对RNA快速成像,并且所报道的染料孵育浓度较高且量子产量也低,而这些高浓度染料可能会影响活细胞的正常生理活动并且与细胞中的背景荧光会产生干扰。重要的是,深红特别是近红外发射的RNA染料比较急需,这是由于在此发射范围特别是NIR生物学窗口(650-900nm)具有最低限度的光损伤、最低限度的背景荧光干扰、降低的光散射和增强的组织穿透深度。因此,开发出新型的具有快速定位、低的孵育浓度、高量子产量、高的光稳定性和近红外发射的RNA染料具有重要的意义。
相对常用染料(香豆素、萘二酰亚胺、BODIPY、菁染料等)中,基于氧杂蒽的荧光染料由于其独特的性质而备受关注:波长长、光稳定性好、量子产量高、摩尔消光系数大、合成简单、易于修饰等,广泛地应用于生物医学领域。然而由于其发射波长短、pH敏感性强等缺点,限制了其在活体领域的应用。近年报道的新型长波长氧杂蒽类荧光染料,主要是通过扩展电子共扼体系、引入吸电子基团等手段来使其荧光红移。然而使其得到的结构具有更长的发射的同时还具有高的荧光量子产量是非常困难的。
发明内容
本发明要解决的第一个技术问题在于提供一种新型近红外的高量子产率染料。此染料具有良好的生物相容性、低毒性、长的荧光发射波长、优异的化学稳定性以及很高的荧光量子产率。
本发明要解决的第二个技术问题在于提供新型近红外的高量子产率染料的制备方法。
本发明要解决的第三个技术问题在于提供一种新型近红外的高量子产率染料作为近红外的核仁RNA染料的应用。
为解决第一个技术问题,本发明一种新型近红外的高量子产率染料,其特征在于,具有如下结构式I:
R1、R2、R6、R7分别独立的为氢、低级烃基、醚基、取代烷基或酰基;R3、R4、R5、R8、R9、R10分别独立的为氢、低级烃基或卤素;R11为氢、甲基、氰基、或三氟甲基; 为阴离子;
所述R1、R2、R6、R7中低级烃基为直链、支链或环状;所述低级烃基包含1-6个碳原子;
所述醚基中碳原子数为3~6,氧原子数≤2;
所述取代烷基为直链或支链;所述取代烷基为ω-甲酸酯基取代1-6个碳原子烷基、ω-甲酰胺基取代1-6个碳原子烷基、ω-卤素取代1-6个碳原子烷基、ω-羟基取代1-6个碳原子烷基、ω-氨基取代1-6个碳原子烷基或ω-巯基取代1-6个碳原子烷基;其中,所述ω-甲酸酯基取代1-6个碳原子烷基中甲酸酯基为2-5个碳原子烷基甲酸酯基:所述的ω-甲酰胺基取代1-6个碳原子烷基中甲酰胺基为2-5个碳原子烃基甲酰胺基;
所述酰基为2-6个碳原子烷基酰基、叔丁氧羰基、苯甲酰基、1-6个碳原子取代苯甲酰基或卤素取代苯甲酰基;
所述卤素为氟、氯、溴或碘;
所述R3、R4、R5、R8、R9、R10中的低级烃基为甲基或乙基;
所述 为平衡电荷的任意有机或无机阴离子。
进一步地,所述结构式I中,R1与R3、R1与R10、R2与R3、R2与R10、R6与R5、R6与R8、R7与R5、R7与R8、R1与R2或R6与R7可以独立形成如下Ia-In结构:
其中R为氢、甲基或乙基;Y为C、O或NR。
更进一步地,所述的R1、R2、R6、R7中的低级烃基为甲基、乙基、丙基、异丙基、环丙基、烯丙基、丁基、异丁基或叔丁基;
所述醚基为CH2CH2OCH3、CH2CH2OCH2CH3、CH2CH2OCH2CH3OH、CH2CH2OCH2CH3OCH3或CH2CH2OCH2CH3OCH2CH3;
所述ω-2-5个碳原子烷基甲酸酯基取代1-6个碳原子烷基为(CH2)mCOO(CH2)nCH3或(CH2)mCOOC(CH3)3,其中m为1-4,n为0-3;
所述ω-2-5个碳原子烃基甲酰胺基取代1-6个碳原子烷基为(CH2)mCONH(CH2)nCH3或(CH2)mCON[(CH2)nCH3]2,其中m为1-4,n为0-3;
所述ω-卤素取代1-6个碳原子烷基为(CH2)mCl、(CH2)mBr或(CH2)mI,其中m为1-6;
所述ω-羟基取代1-6个碳原子烷基为(CH2)mOH,其中m为1-6;
所述ω-氨基取代1-6个碳原子烷基为(CH2)mNH2,其中m为1-6;
所述ω-巯基取代1-6个碳原子烷基为(CH2)mSH,其中m为1-6;
所述2-6个碳原子烷基酰基为乙酰基、丙酰基、丁酰基或叔丁酰基;
所述1-6个碳原子取代苯甲酰基为甲基取代苯甲酰基、乙基取代苯甲酰基、丙基取代苯甲酰基、丁基取代苯甲酰基或叔丁基取代苯甲酰基;
所述卤素取代苯甲酰基为氯取代苯甲酰基、溴取代苯甲酰基或碘取代苯甲酰基。
为解决第二个技术问题,本发明提供新型近红外的高量子产率染料的制备方法,当结构式I中R11为氢或三氟甲基时,包括如下制备步骤:
将1毫摩尔化合物II与1毫摩尔化合物III加入到5-15毫升浓硫酸中,在加热条件下反应1-4小时,冷却后将反应液慢慢滴加20-100倍体积的冰水中,再加入0.01-1倍体积的质量百分浓度为70%的高氯酸,充分搅拌,析出固体,过滤、真空干燥,柱层析分离得产物。
其中,所述加热的温度为60-100℃;
所述化合物II的结构式如下:
所述化合物III的结构式如下:
其中,R1、R2、R6、R7分别独立的为氢、低级烃基、醚基、取代烷基或酰基;R3、R4、R5、R8、R9、R10分别独立的为氢、低级烃基或卤素;R0为氢或三氟甲基。
或,当结构式I中R11为甲基时,包括如下制备步骤:
1)将1毫摩尔化合物II与1毫摩尔化合物III加入到5-15毫升浓硫酸中,在加热条件下反应1-4小时,冷却后将反应液慢慢滴加20-100倍体积的冰水中,再加入0.01-1倍体积的质量百分浓度为70%的高氯酸,充分搅拌,析出固体,过滤、真空干燥,柱层析分离得产物1-a;
2)将1毫摩尔得到的I-a溶于5毫升无水THF中,在N2保护条件下逐滴加入3-6毫升中含有1毫摩尔CH3MgI的THF溶液,常温搅拌24h后,再加入2-5毫升含有1毫摩尔DDQ的THF溶液,继续搅拌1-12h,将反应液旋干后柱层析分离得到产物。
其中,所述加热的温度为60-100℃;
所述化合物II的结构式如下:
所述化合物III的结构式如下:
其中,R1、R2、R6、R7分别独立的为氢、低级烃基、醚基、取代烷基或酰基;R3、R4、R5、R8、R9、R10分别独立的为氢、低级烃基或卤素;R0为氢。
或,当结构式I中R11为氰基时,包括如下制备步骤:
①将1毫摩尔化合物II与1毫摩尔化合物III加入到5-15毫升浓硫酸中,在加热条件下反应1-4小时,冷却后将反应液慢慢滴加20-100倍体积的冰水中,再加入0.01-1倍体积的质量百分浓度为70%的高氯酸,充分搅拌,析出固体,过滤、真空干燥,柱层析分离得产物1-a;
②将1毫摩尔得到的I-a溶于5-20毫升无水DMF中,再加入1毫摩尔的NaCN,常温搅拌24h后,再加入2-5毫升中含有1毫摩尔DDQ的THF溶液,将反应液旋干后柱层析分离得到产物。
其中,所述加热的温度为60-100℃;
所述化合物II的结构式如下:
所述化合物III的结构式如下:
其中,R1、R2、R6、R7分别独立的为氢、低级烃基、醚基、取代烷基或酰基;R3、R4、R5、R8、R9、R10分别独立的为氢、低级烃基或卤素;R0为氢。
进一步地,上述三种制备方法中,所述R1、R2、R6、R7中低级烃基为直链、支链或环状;所述低级烃基包含1-6个碳原子;
所述醚基中碳原子数为3-6,氧原子数≤2;
所述取代烷基为直链或支链;所述取代烷基为ω-甲酸酯基取代1-6个碳原子烷基、ω-甲酰胺基取代1-6个碳原子烷基、ω-卤素取代1-6个碳原子烷基、ω-羟基取代1-6个碳原子烷基、ω-氨基取代1-6个碳原子烷基或ω-巯基取代1-6个碳原子烷基;其中所述ω-甲酸酯基取代1-6个碳原子烷基中甲酸酯基为2-5个碳原子烷基甲酸酯基;所述的ω-甲酰胺基取代1-6个碳原子烷基中甲酰胺基为2-5个碳原子烃基甲酰胺基;
所述酰基为2-6个碳原子烷基酰基、叔丁氧羰基、苯甲酰基、1-6个碳原子取代苯甲酰基或卤素取代苯甲酰基;
所述卤素为氟、氯、溴或碘;
所述R3、R4、R5、R8、R9、R10中的低级烃基为甲基或乙基。
进一步地,所述化合物II和化合物III可以独立形成如下IIa-IIg和IIIa-IIIg结构:
其中R为氢、甲基或乙基;Y为C、O或NR。
优选地,所述R1、R2、R6、R7中的低级烃基为甲基、乙基、丙基、异丙基、环丙基、烯丙基、丁基、异丁基或叔丁基;
优选地,所述醚基为CH2CH2OCH3、CH2CH2OCH2CH3、CH2CH2OCH2CH3OH、CH2CH2OCH2CH3OCH3或CH2CH2OCH2CH3OCH2CH3;
优选地,所述ω-2-5个碳原子烷基甲酸酯基取代1-6个碳原子烷基为(CH2)mCOO(CH2)nCH3或(CH2)mCOOC(CH3)3,其中m为1-4,n为0-3;
优选地,所述ω-2-5个碳原子烃基甲酰胺基取代1-6个碳原子烷基为(CH2)mCONH(CH2)nCH3或(CH2)mCON[(CH2)nCH3]2,其中m为1-4,n为0-3;
优选地,所述ω-卤素取代1-6个碳原子烷基为(CH2)mCl、(CH2)mBr或(CH2)mI,其中m为1-6;
优选地,所述ω-羟基取代1-6个碳原子烷基为(CH2)mOH,其中m为1-6;
优选地,所述ω-氨基取代1-6个碳原子烷基为(CH2)mNH2,其中m为1-6;
优选地,所述ω-巯基取代1-6个碳原子烷基为(CH2)mSH,其中m为1-6;
优选地,所述2-6个碳原子烷基酰基为乙酰基、丙酰基、丁酰基或叔丁酰基;
优选地,所述1-6个碳原子取代苯甲酰基为甲基取代苯甲酰基、乙基取代苯甲酰基、丙基取代苯甲酰基、丁基取代苯甲酰基或叔丁基取代苯甲酰基;
优选地,所述卤素取代苯甲酰基为氯取代苯甲酰基、溴取代苯甲酰基或碘取代苯甲酰基。
为解决第三个技术问题,本发明保护新型近红外的高量子产率染料作为近红外的核仁RNA染料的应用。
进一步地,本发明保护新型近红外的高量子产率染料作为近红外的核仁RNA染料,应用于核仁RNA的荧光标记。
一些新的抗癌药物,如顺铂、放线菌素D、α-鹅膏蕈碱等的作用位点是RNA聚合酶,从而诱导细胞的死亡,而RNA聚合酶中,RNA聚合酶I和RNA聚合酶III位于细胞质中,RNA聚合酶II位于细胞核中,因此本发明中的染料通过对核仁RNA进行荧光标记,可以用来筛选不同作用位点的抗癌药物以及对抗癌药物药效的评估。
本发明基于原料易得的3,4-二氢-1-萘酮衍生物,通过与其对应的水杨醛衍生物在浓硫酸的缩合条件下加热,得到了一系列不对称、非直线型共轭结构的新型氧杂蒽衍生的化合物,此化合物具有较长的荧光发射(近红外荧光),特别是结构中引入的亚甲基结构使其具有较高的荧光量子产率。此外其良好的生物相容性和低毒性,可以对生物体系的大分子(核仁RNA)进行荧光标记,对研究其在生理活性中的作用奠定了重要的基础。
本发明的有益效果如下:
本发明的具有结构式I的新型近红外的高量子产率染料具有不对称、非直线型的共轭结构,并且具有长的荧光发射(近红外发射)和较高的荧光量子产率,同时也具有良好的生物相容性和低毒性,可以对生物体系尤其是细胞中的重要生物大分子核仁RNA荧光标记,降低了染料孵育浓度,避免了高浓度染料对细胞正常生理功能的干扰,同时避免了背景荧光的干扰从而对生物大分子准确的定位,对于研究核仁RNA相关的生理学过程有着重要的作用。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细的说明。
图1示出本发明实例3中荧光染料I-6将A549细胞染色后的图。
图2示出本发明实例7中荧光染料I-15将A549细胞染色后的图。
图3示出本发明实例15中荧光染料I-29将A549细胞染色后的图。
图4示出本发明实例16中荧光染料I-30将A549细胞染色后的图。
图5示出本发明实例27中荧光染料I-42将A549细胞染色后的图。
图6示出本发明实例28中荧光染料I-43将A549细胞染色后的图。
图7示出本发明实例37中荧光染料I-54将A549细胞染色后的图。
图8示出本发明实例37中荧光染料I-54将HeLa细胞染色后的图。
图9示出本发明实例40中荧光染料I-78将A549细胞染色后的图。
图10示出本发明实例40中荧光染料I-78将HeLa细胞染色后的图。
图11示出本发明实例42中荧光染料I-95将HeLa细胞染色后的图。
图12示出本发明实例54中荧光染料I-191将HeLa细胞染色后的图。
图13示出本发明实例62中荧光染料I-255将HeLa细胞染色后的图。
图14示出本发明实例77中荧光染料I-393将HeLa细胞染色后的图。
图15示出本发明实例37的荧光染料I-54和实例40中的荧光染料I-78分别和I-78加DNA酶和RNA酶消化后并与市售DNA、RNA染料对比图。
图16示出本发明实例37的荧光染料I-54和实例40中的荧光染料I-78分别与市售RNA染料SYTO RNASelect的细胞染色效果对比图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例和附图对本发明做进一步的说明。附图中相似的部件以相同的附图标记进行表示。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
实施例1:
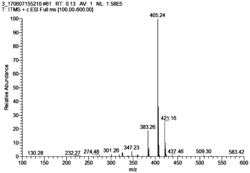
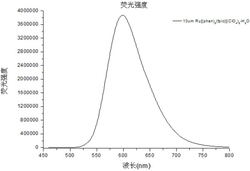
将0.193g(0.001mol)的化合物II-1和0.161g(0.001mol)的化合物III-1分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.343g化合物I-1,产率82%。ESI MS:理论计算m/z:319.18,实际测试m/z:319.20。λabs.max/nm=615nm,λemmax/nm=638nm,Фf=0.80。
将0.418g(0.001mol)的化合物I-1、0.202g(0.001mol)的二碳酸二叔丁酯和0.101g(0.001mol)的三乙胺分别加入到10毫升的无水二氯甲烷中,常温条件下搅拌24h,在真空条件下除去溶剂,干燥,柱层析纯化分离得到0.440g化合物I-2,产率85%。ESI MS:理论计算m/z:419.23,实际测试m/z:419.25。λabs.max/nm=620nm,λemmax/nm=645nm,Фf=0.82。
将0.518g(0.001mmol)化合物I-2溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.223g化合物I-3,产率42%。ESI MS:理论计算m/z:433.25,实际测试m/z:433.20。λabs.max/nm=622nm,λemmax/nm=648nm,Фf=0.85。
实施例2
化合物III-11根据文献(Org.Lett.,2011,13,6488-6491)由商用化合物III-1与碘乙烷制备,产率50%。
将0.217g(0.001mol)的化合物II-2和0.189g(0.001mol)的化合物III-2分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.399g化合物I-4,产率85%。ESI MS:理论计算m/z:371.21,实际测试m/z:371.24。λabs.max/nm=635nm,λemmax/nm=658nm,Фf=0.73。
将0.470g(0.001mmol)化合物I-4溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.198g化合物I-5,产率41%。ESI MS:理论计算m/z:385.23,实际测试m/z:383.27。λabs.max/nm=636nm,λemmax/nm=660nm,Фf=0.75。
实施例3
化合物II-3根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由7-羟基-1-甲基-1,2,3,4-四氢喹啉制备,产率76%。化合物III-3根据文献(Org.Lett.,2011,13,6488–6491)由商用化合物III-1与碘甲烷制备,产率42%。
将0.191g(0.001mol)的化合物II-3和0.189g(0.001mol)的化合物III-3分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.391g化合物I-6,产率88%。ESI MS:理论计算m/z:345.20,实际测试m/z:345.26。λabs.max/nm=633nm,λemmax/nm=656nm,Фf=0.78。
将0.444g(0.001mmol)化合物I-6溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.202g化合物I-7,产率44%。ESI MS:理论计算m/z:359.21,实际测试m/z:359.17。λabs.max/nm=634nm,λemmax/nm=658nm,Фf=0.80。
实施例4
化合物II-4根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由7-羟基-1-乙基-2,2,4-三甲基-1,2-二氢喹啉制备,产率71%。化合物III-4根据文献(Org.Lett.,2011,13,6488–6491)由商用化合物III-1与1-碘乙烷制备,产率45%。
将0.231g(0.001mol)的化合物II-4和0.217g(0.001mol)的化合物III-4分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.446g化合物I-8,产率87%。ESI MS:理论计算m/z:413.26,实际测试m/z:413.32。λabs.max/nm=648nm,λemmax/nm=674nm,Фf=0.76。
将0.512g(0.001mmol)化合物I-8溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.226g化合物I-9,产率43%。ESI MS:理论计算m/z:359.21,实际测试m/z:359.17。λabs.max/nm=650nm,λemmax/nm=678nm,Фf=0.73。
实施例5
化合物II-5根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由3-N,N-二乙氨基苯酚制备,产率75%。化合物III-5根据文献(Org.Lett.,2011,13,6488–6491)由商用化合物III-1与1-溴-2-(2-甲氧基乙氧基)乙烷制备,产率30%。
将0.165g(0.001mol)的化合物II-5和0.365g(0.001mol)的化合物III-5分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.440g化合物I-10,产率74%。ESI MS:理论计算m/z:495.29,实际测试m/z:495.23。λabs.max/nm=622nm,λemmax/nm=648nm,Фf=0.80。
将0.594g(0.001mmol)化合物I-10溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.255g化合物I-11,产率42%。ESI MS:理论计算m/z:509.30,实际测试m/z:509.27。λabs.max/nm=623nm,λemmax/nm=650nm,Фf=0.83。
实施例6
化合物II-6根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由7-羟基-1-(4-正丁酸)-1,2,3,4-四氢喹啉制备,产率62%。化合物III-6根据文献(Org.Lett.,2011,13,6488–6491)由商用化合物III-1分别与1-氯-3-溴丙烷、碘甲烷制备,产率28%。
将0.291g(0.001mol)的化合物II-6和0.215g(0.001mol)的化合物III-6分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.393g化合物I-12,产率69%。ESI MS:理论计算m/z:471.26,实际测试m/z:471.21。λabs.max/nm=632nm,λemmax/nm=658nm,Фf=0.74。
将0.570g(0.001mmol)化合物I-12溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.222g化合物I-13,产率38%。ESI MS:理论计算m/z:485.28,实际测试m/z:485.32。λabs.max/nm=633nm,λemmax/nm=662nm,Фf=0.76。
实施例7
化合物II-7根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由3-(1-哌啶基)苯酚制备,产率83%。化合物III-7根据文献(Org.Lett.,2011,13,6488–6491)由商用化合物III-1与1-氯-3-溴丙烷制备,产率20%。
将0.205g(0.001mol)的化合物II-7和0.241g(0.001mol)的化合物III-7分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.454g化合物I-14,产率89%。ESI MS:理论计算m/z:411.24,实际测试m/z:411.20。λabs.max/nm=630nm,λemmax/nm=656nm,Фf=0.74。
将0.510g(0.001mmol)化合物I-14溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.220g化合物I-15,产率42%。ESI MS:理论计算m/z:425.26,实际测试m/z:425.22。λabs.max/nm=630nm,λemmax/nm=660nm,Фf=0.75。
实施例8
化合物II-8根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由3-(4-吗啉基)苯酚制备,产率83%。化合物III-8根据文献(Org.Lett.,2011,13,6488–6491)由商用化合物III-1与1,5-二溴戊烷制备,产率14%。
将0.207g(0.001mol)的化合物II-8和0.229g(0.001mol)的化合物III-8分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.400g化合物I-16,产率80%。ESI MS:理论计算m/z:401.22,实际测试m/z:401.20。λabs.max/nm=624nm,λemmax/nm=652nm,Фf=0.83。
将0.500g(0.001mmol)化合物I-16溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.200g化合物I-17,产率39%。ESI MS:理论计算m/z:415.24,实际测试m/z:415.27。λabs.max/nm=621nm,λemmax/nm=654nm,Фf=0.84。
实施例9
化合物II-9根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由3-(1-哌嗪)苯酚制备,产率68%。化合物III-9根据文献(Org.Lett.,2011,13,6488–6491)由商用化合物III-1与1-溴-2-(2-溴乙氧基)乙烷制备,产率11%。
将0.206g(0.001mol)的化合物II-9和0.231g(0.001mol)的化合物III-9分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.381g化合物I-18,产率76%。ESI MS:理论计算m/z:402.22,实际测试m/z:402.25。λabs.max/nm=625nm,λemmax/nm=653nm,Фf=0.85。
将0.501g(0.001mmol)化合物I-18溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.191g化合物I-19,产率37%。ESI MS:理论计算m/z:416.23,实际测试m/z:416.19。λabs.max/nm=624nm,λemmax/nm=655nm,Фf=0.87。
实施例10
化合物II-10根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由间氨基苯酚制备,产率48%。
将0.137g(0.001mol)的化合物II-10和0.189g(0.001mol)的化合物III-3分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.304g化合物I-20,产率78%。ESI MS:理论计算m/z:291.15,实际测试m/z:291.20。λabs.max/nm=617nm,λemmax/nm=636nm,Фf=0.82。
将0.390g(0.001mol)的化合物I-20、0.202g(0.001mol)的二碳酸二叔丁酯和0.101g(0.001mol)的三乙胺分别加入到10毫升的无水二氯甲烷中,常温条件下搅拌24h,在真空条件下除去溶剂,干燥,柱层析纯化分离得到0.392g化合物I-21,产率80%。ESI MS:理论计算m/z:391.20,实际测试m/z:391.25。λabs.max/nm=620nm,λemmax/nm=644nm,Фf=0.85。
将0.490g(0.001mmol)化合物I-21溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.186g化合物I-22,产率37%。ESI MS:理论计算m/z:405.22,实际测试m/z:405.24。λabs.max/nm=620nm,λemmax/nm=646nm,Фf=0.87。
实施例11
化合物II-10根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由3-乙氨基-4-甲基苯酚制备,产率56%。
将0.179g(0.001mol)的化合物II-11和0.217g(0.001mol)的化合物III-4分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.359g化合物I-23,产率78%。ESI MS:理论计算m/z:361.23,实际测试m/z:361.18。λabs.max/nm=623nm,λemmax/nm=648nm,Фf=0.87。
将0.460g(0.001mmol)化合物I-23溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.166g化合物I-24,产率35%。ESI MS:理论计算m/z:375.24,实际测试m/z:375.29。λabs.max/nm=621nm,λemmax/nm=650nm,Фf=0.89。
实施例12
化合物II-12根据文献(J.Am.Chem.Soc.,2007,129,9986-9998)由4-氯-3-二乙胺基苯酚制备,产率56%;化合物III-10根据文献(Org.Lett.,2011,13,6488–6491)由商用化合物III-1与双(2-溴乙基)胺制备,产率8%。
将0.227g(0.001mol)的化合物II-11和0.230g(0.001mol)的化合物III-4分别加入到5毫升的浓硫酸中,然后在100℃条件下加热搅拌,反应2h。冷却后,将反应液缓慢倒入到200毫升冰水中,然后在搅拌的条件下逐滴加入1毫升高氯酸(70%),再加入大量的蒸馏水,静置后析出固体,过滤、真空干燥,柱层析纯化分离得到0.375g化合物I-25,产率72%。ESI MS:理论计算m/z:422.20,实际测试m/z:422.23。λabs.max/nm=624nm,λemmax/nm=652nm,Фf=0.83。
将0.521g(0.001mmol)化合物I-25溶于5毫升无水THF,氮气保护下逐滴加入3毫升中含有1毫摩尔CH3MgI的THF溶液,常温反应24小时,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.176g化合物I-26,产率33%。ESI MS:理论计算m/z:436.22,实际测试m/z:436.19。λabs.max/nm=622nm,λemmax/nm=655nm,Фf=0.85。
实施例13
将0.518g(0.001mmol)化合物I-2溶于5毫升无水DMF,然后加入1毫摩尔的NaCN,常温搅拌24h后,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.196g化合物I-27,产率36%。ESI MS:理论计算m/z:444.23,实际测试m/z:444.21。λabs.max/nm=690nm,λemmax/nm=720nm,Фf=0.35。
实施例14
将0.470g(0.001mmol)化合物I-4溶于5毫升无水DMF,然后加入1毫摩尔的NaCN,常温搅拌24h后,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.198g化合物I-28,产率40%。ESI MS:理论计算m/z:396.21,实际测试m/z:396.23。λabs.max/nm=698nm,λemmax/nm=732nm,Фf=0.25。
实施例15
将0.444g(0.001mmol)化合物I-6溶于5毫升无水DMF,然后加入1毫摩尔的NaCN,常温搅拌24h后,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.197g化合物I-29,产率42%。ESI MS:理论计算m/z:370.19,实际测试m/z:370.17。λabs.max/nm=705nm,λemmax/nm=730nm,Фf=0.30。
实施例16
将0.512g(0.001mmol)化合物I-8溶于5毫升无水DMF,然后加入1毫摩尔的NaCN,常温搅拌24h后,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.236g化合物I-30,产率44%。ESI MS:理论计算m/z:438.25,实际测试m/z:438.29。λabs.max/nm=715nm,λemmax/nm=750nm,Фf=0.20。
实施例17
将0.594g(0.001mmol)化合物I-10溶于5毫升无水DMF,然后加入1毫摩尔的NaCN,常温搅拌24h后,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.254g化合物I-31,产率41%。ESI MS:理论计算m/z:520.28,实际测试m/z:520.30。λabs.max/nm=694nm,λemmax/nm=612nm,Фf=0.33。
实施例18
将0.570g(0.001mmol)化合物I-12溶于5毫升无水DMF,然后加入1毫摩尔的NaCN,常温搅拌24h后,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.226g化合物I-32,产率38%。ESI MS:理论计算m/z:496.26,实际测试m/z:496.30。λabs.max/nm=706nm,λemmax/nm=734nm,Фf=0.26。
实施例19
将0.510g(0.001mmol)化合物I-14溶于5毫升无水DMF,然后加入1毫摩尔的NaCN,常温搅拌24h后,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.235g化合物I-33,产率44%。ESI MS:理论计算m/z:436.24,实际测试m/z:436.22。λabs.max/nm=705nm,λemmax/nm=735nm,Фf=0.25。
实施例20
将0.500g(0.001mmol)化合物I-16溶于5毫升无水DMF,然后加入1毫摩尔的NaCN,常温搅拌24h后,再加入2毫升中含有1毫摩尔DDQ的THF溶液,继续搅拌1h,在真空条件下除去溶剂,干燥,柱层析分离得到0.221g化合物I-34,产率42%。ESI MS:理论计算m/z:426.22,实际测试m/z:426.27。λabs.max/nm=694nm,λemmax/nm
一种近红外的高量子产率染料及其制备与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0