








专利摘要
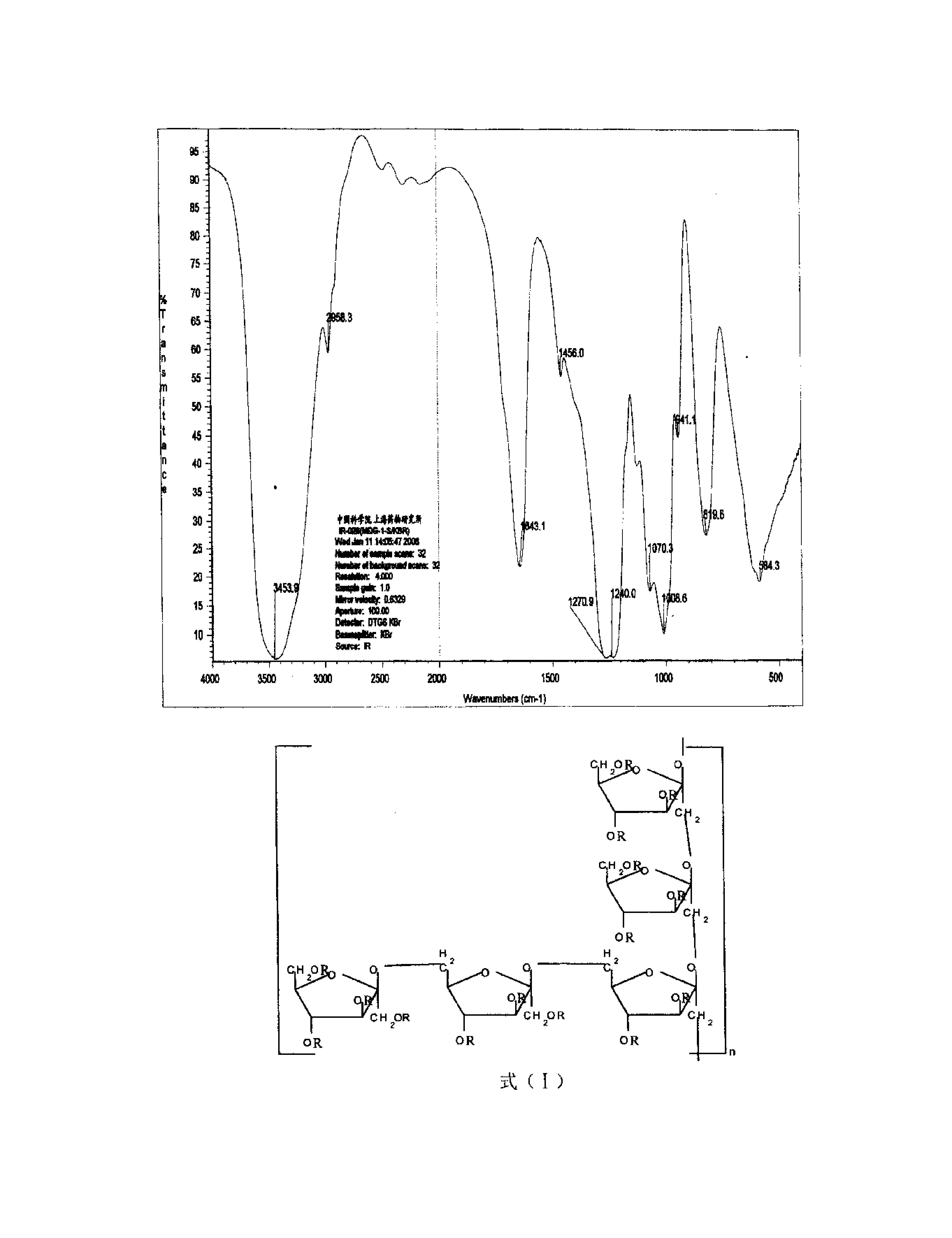
本发明提供一种与褐藻Sargassum?polycystum多糖相关的硫酸化岩藻-半乳四糖及其制备方法,以L-岩藻糖和保护的D-半乳糖中间体为原料,分别合成四个单糖片段,通过汇聚式组装,将两个单糖化合物合成二糖化合物,再将两个二糖化合物合成四糖化合物,经脱酰基保护、硫酸化、脱苄基后到式(I)硫酸化岩藻-半乳四糖。本发明硫酸化岩藻-半乳四糖具有抑制HSV-1感染的活性,具有作为抑制HSV-1感染的药物的应用前景。
权利要求
1.一种硫酸化岩藻-半乳四糖,其特征在于,其化学结构如式(I)所示,其化学结构简式如式(II)所示:
2.一种硫酸化岩藻-半乳四糖的制备方法,其特征在于,以L-岩藻糖和保护的D-半乳糖中间体为原料分别合成四个单糖片段,通过汇聚式组装,将两个单糖化合物合成二糖化合物,再将两个二糖化合物合成四糖化合物,经脱酰基保护、硫酸化、脱苄基后到式(I)硫酸化岩藻-半乳四糖;所述制备方法包括以下步骤及反应式:
步骤(1):L-岩藻糖经过全乙酰化得到化合物1;通过4-甲基苯硫酚进行硫苷化得到硫苷2;再经过乙酰基脱除、3,4-O-异亚丙基保护、2-O-苄基化等3步得到3;然后,经酸性条件脱除3,4-O-异亚丙基保护,得到3,4-二醇4;采用锡缩酮活化的方式进行3-O-苄基化而得到5;再采用苯甲酰基保护得到6;中间体6中经过含卤素氧化剂处理,得到化合物7;再用三氯乙腈、碱处理得到8,或用N-苯基三氟乙酰亚胺酰氯、碱处理得到化合物9;
步骤(2):化合物2经过乙酰基脱除、3,4-O-原苯甲酸酯保护、2-O-苄基化保护、酸性条件下水解后得到化合物10;
步骤(3):化合物1经过酚苷化得到中间体11;再经过乙酰基脱除、3,4-O-原苯甲酸酯保护、2-O-苄基化保护、酸性条件下水解后得到化合物12;
步骤(4):化合物13经过酸性条件下脱除苄缩醛保护,得到中间体14;再经过苄溴/碱/季铵盐在二氯甲烷和水的混合溶剂中处理,得到中间体15;然后经过乙酰化得到化合物16;
步骤(5):将所述化合物10和所述化合物8或化合物9在糖基化条件下反应,得到二糖中间体化合物17
步骤(6):所述化合物16和化合物12在糖基化条件下得到二糖中间体18;再经过DDQ脱除4-甲氧基苄基保护,得到中间体19;
步骤(7):所述化合物19和化合物17在糖基化条件下得到四糖中间体20;
步骤(8):化合物20经过碱处理得到四醇21;经硫酸化试剂处理,得到硫酸化中间体22;再在催化剂条件下氢解脱苄,得到式(I)硫酸化岩藻-半乳四糖;
3.如权利要求2所述的制备方法,其特征在于,所述步骤(1)中,所述含卤素氧化剂为N-碘代琥珀酰亚胺、N-溴代琥珀酰亚胺、乙酸碘苯;所述碱为:碳酸钾、碳酸铯、碳酸钠、1,8-二氮杂二环十一碳-7-烯、氢化钠;所述化合物9的异头碳上的构型为β-构型,α-构型或α、β构型的混合物。
4.如权利要求2所述的制备方法,其特征在于,所述步骤(4)中,所述碱为碳酸钠、碳酸钾、氢氧化钠、氢氧化钾;所述季铵盐为四丁基硫酸氢铵、四丁基溴化铵、四乙基氯化铵、十二烷基三甲基溴化铵;所述二氯甲烷和水的体积比为1∶4~4∶1。
5.如权利要求2所述的制备方法,其特征在于,所述步骤(5)中,所述糖基化条件为路易斯酸或质子酸、溶剂、温度的组合;其中,所述路易斯酸为三甲基硅基三氟甲磺酸酯、叔丁基二甲基硅基三氟甲磺酸酯、三乙基硅基三氟甲磺酸酯、三氟化硼乙醚、三氟甲磺酸银;所述质子酸为三氟甲磺酸、三氟乙酸;所述溶剂为乙醚、二氯甲烷、1,4-二氧六环、四氢呋喃或其二元混合物、三元混合物;所述温度为-78℃~-20℃。
6.如权利要求2所述的制备方法,其特征在于,所述步骤(6)中,所述糖基化条件为含卤素氧化剂、路易斯酸或质子酸、溶剂、温度的组合;其中,所述含卤素氧化剂为:N-碘代琥珀酰亚胺、N-溴代琥珀酰亚胺、乙酸碘苯;所述路易斯酸为三甲基硅基三氟甲磺酸酯、叔丁基二甲基硅基三氟甲磺酸酯、三乙基硅基三氟甲磺酸酯、三氟化硼乙醚、三氟甲磺酸银;所述质子酸为三氟甲磺酸、三氟乙酸;所述溶剂为乙醚、二氯甲烷、1,4-二氧六环、四氢呋喃或其二元混合物、三元混合物;所述温度为-40℃~0℃。
7.如权利要求2所述的制备方法,其特征在于,所述步骤(7)中,糖基化条件为含卤素氧化剂、路易斯酸或质子酸、溶剂、温度的组合;其中,卤素氧化剂为N-碘代琥珀酰亚胺、N-溴代琥珀酰亚胺、乙酸碘苯;路易斯酸为三甲基硅基三氟甲磺酸酯、叔丁基二甲基硅基三氟甲磺酸酯、三乙基硅基三氟甲磺酸酯、三氟化硼乙醚、三氟甲磺酸银;质子酸为:三氟甲磺酸、三氟乙酸;溶剂为:乙醚、二氯甲烷、1,4-二氧六环、四氢呋喃或其二元混合物、三元混合物;温度为:-20℃~0℃。
8.如权利要求2所述的制备方法,其特征在于,所述步骤(8)中,所述碱为甲醇钠、乙醇钠、碳酸钠、碳酸钾;所述硫酸化试剂为三氧化硫-吡啶络合物、三氧化硫-三甲胺络合物、三氧化硫-三乙胺络合物;所述催化剂为钯/炭、氢氧化钯/炭、钯黑、铂/炭。
9.式(I)硫酸化岩藻-半乳四糖在制备治疗HSV-1感染疾病的药物中的应用,其特征在于,所述式(I)硫酸化岩藻-半乳四糖抑制HSV-1感染。
说明书
技术领域
本发明属于有机合成、天然产物化学等技术领域。具体涉及一种褐藻硫酸化岩藻-半乳四糖及其制备方法和应用。
背景技术
随着海洋科学的发展,海洋天然多糖逐渐得到开发和广泛关注,并应用于医药、生命科学研究。岩藻多糖(fucoidans)是广泛存在于海洋生物褐藻(brown alga)和一些海洋无脊椎动物中的一类复杂硫酸化水溶性多糖。这类硫酸化岩藻多糖在研究中体现出了广泛的生理和生物学功能,比如:双向调节免疫力,清除自由基,抗衰老,抗凝血和抗血栓,抗肿瘤和HIV病毒,消除胃肠系统紊乱,抗过敏,增强肝功能,降低高血脂和高血压等功效。低分子量岩藻多糖具有:抗凝血、抗病毒、抗血栓形成、抗肿瘤、抗炎和增强免疫力等活性。
这类硫酸化岩藻多糖都具有共同的化学结构特点,即:由硫酸化的α-L-吡喃岩藻糖(Fucp)残基通过(1→3)-或者(1→4)-连接的而成的多糖(如式XI所示)。
然而,根据海藻种类和来源的不同,粗提取的岩藻多糖中还含有由不同糖残基经各种连接方式组成的杂多糖。例如,在2013年,Usov课题组从褐藻Sargassum polycystum中提取出了一种新的半乳-岩藻多糖,该多糖具有一种罕见的糖残基序列(如式XII所示),即:[→3)-α-L-Fucp4S-(1→3)-α-L-Fucp4S-(1→2)-α-D-Galp4S-(1→3)-α-L-Fucp4S-(1→]重复单元。其中的[→2)-α-D-Galp-(1→]单元是一种在天然海藻来源中很不寻常的结构特征,因此,将对分子构象及生理活性具有一定的影响。
式XII所述的硫酸化岩藻多糖属于高分子量的多糖分子,其聚合度n的不确定性以及过大的分子量都限制了其在生物活性、构效关系以及药物开发中的研究和应用。因而,人们希望获得该岩藻多糖相应的、低分子量、低聚合度的寡糖分子(例如:聚合度n=1,2.....),以研究其生理活性。然而,化学降解或者酶降解法都难以得到结构确定的低分子量寡糖。因此,相应的低分子量寡糖必须通过化学合成的方法获得。迄今为止,该硫酸化岩藻多糖对应的低分子量寡糖分子还未曾见文献报道。
发明内容
本发明提供一种与褐藻Sargassum polycystum多糖相关的硫酸化岩藻-半乳四糖。该四糖残基序列为:α-L-Fucp4S-(1→3)-α-L-Fucp4S-(1→2)-α-D-Galp4S-(1→3)-α-L-Fucp4S-(1→OPMP。该四糖结构中,各个糖残基的4-O-位均为硫酸化,四个糖苷键均为α-构型。其化学结构式(I)和化学结构简式(II)分别如下:
本发明还提供了一种硫酸化岩藻-半乳四糖的制备方法,以L-岩藻糖和保护的D-半乳糖中间体为原料,分别合成四个单糖片段,通过汇聚式组装,将两个单糖化合物合成二糖化合物,再将两个二糖化合物合成四糖化合物,经脱酰基保护、硫酸化、脱苄基后到式(I)硫酸化岩藻-半乳四糖;
所述制备方法包括以下步骤及反应式:
步骤(1):L-岩藻糖经过全乙酰化得到化合物1;通过4-甲基苯硫酚进行硫苷化得到硫苷2;再经过乙酰基脱除、3,4-O-异亚丙基保护、2-O-苄基化等3步得到3;然后,经酸性条件脱除3,4-O-异亚丙基保护,得到3,4-二醇4;采用锡缩酮活化的方式进行3-O-苄基化而得到5;再采用苯甲酰基保护得到6;中间体6中经过含卤素氧化剂处理,得到化合物7;再用三氯乙腈、碱处理得到8,或用N-苯基三氟乙酰亚胺酰氯、碱处理得到化合物9;
步骤(2):化合物2经过乙酰基脱除、3,4-O-原苯甲酸酯保护、2-O-苄基化保护、酸性条件下水解后得到化合物10;
步骤(3):化合物1经过酚苷化得到中间体11;再经过乙酰基脱除、3,4-O-原苯甲酸酯保护、2-O-苄基化保护、酸性条件下水解后得到化合物12;
步骤(4):化合物13经过酸性条件下脱除苄缩醛保护,得到中间体14;再经过苄溴/碱/季铵盐在二氯甲烷和水的混合溶剂中处理,得到中间体15;然后经过乙酰化得到化合物16;
步骤(5):将所述化合物10和所述化合物8或化合物9在糖基化条件下反应,得到二糖中间体化合物17
步骤(6):所述化合物16和化合物12在糖基化条件下得到二糖中间体18;再经过DDQ脱除4-甲氧基苄基保护,得到中间体19;
步骤(7):所述化合物19和化合物17在糖基化条件下得到四糖中间体20;
步骤(8):化合物20经过碱处理得到四醇21;经硫酸化试剂处理,得到硫酸化中间体22;再在催化剂条件下氢解脱苄,得到式(I)硫酸化岩藻-半乳四糖;
本发明中,如式(III)所示的化合物8和9的合成。具体步骤为:L-岩藻糖,经过全乙酰化得到1;通过4-甲基苯硫酚进行硫苷化得到硫苷2;再经过乙酰基脱除、3,4-O-异亚丙基保护、2-O-苄基化等3步得到3;然后,经酸性条件脱除3,4-O-异亚丙基保护,得到3,4-二醇4;采用锡缩酮活化的方式进行3-O-苄基化而得到5;再采用苯甲酰基保护得到6;中间体6经过含卤素氧化剂处理,得到化合物7;再用三氯乙腈、碱处理得到8,或用N-苯基三氟乙酰亚胺酰氯、碱处理得到9。
其中,含卤素氧化剂为:N-碘代琥珀酰亚胺、N-溴代琥珀酰亚胺、乙酸碘苯等。碱为:碳酸钾、碳酸铯、碳酸钠、1,8-二氮杂二环十一碳-7-烯、氢化钠等。如式(III)中所述的化合物9,异头碳上的构型可为:β-构型,α-构型或者α、β构型的混合物。
本发明中,如式(IV)所示的化合物10的合成。具体步骤为:中间体2,经过乙酰基脱除、3,4-O-原苯甲酸酯保护、2-O-苄基化保护、酸性条件下水解等4步得到10。
本发明中,如式(V)所示的化合物12的合成。具体步骤为:中间体1,经过酚苷化得到中间体11;再经过乙酰基脱除、3,4-O-原苯甲酸酯保护、2-O-苄基化保护、酸性条件下水解等4步得到12。
本发明中,如式(VI)所示,化合物16的合成。具体步骤为:化合物13经过酸性条件下脱除苄缩醛保护,得到中间体14;再经过苄溴/碱/季铵盐在二氯甲烷和水的混合溶剂中处理,得到中间体15;然后经过乙酰化得到16。
其中,碱为:碳酸钠、碳酸钾、氢氧化钠、氢氧化钾等。其中,季铵盐为:四丁基硫酸氢铵、四丁基溴化铵、四乙基氯化铵、十二烷基三甲基溴化铵等。其中,二氯甲烷和水的体积比为:1∶4~4∶1。
本发明中,如式(VII)所示,化合物10和化合物8或者化合物9,在糖基化条件下得到二糖中间体17。
其中,糖基化条件为路易斯酸(或质子酸)、溶剂、温度的组合。其中,路易斯酸为:三甲基硅基三氟甲磺酸酯、叔丁基二甲基硅基三氟甲磺酸酯、三乙基硅基三氟甲磺酸酯、三氟化硼乙醚、三氟甲磺酸银等。质子酸为:三氟甲磺酸、三氟乙酸等。溶剂为:乙醚、二氯甲烷、1,4-二氧六环、四氢呋喃或其二元混合物、三元混合物。温度为:-78℃~-20℃。
本发明中,如式(VIII)所示,化合物16和化合物12,在糖基化条件下得到二糖中间体18;再经过DDQ脱除4-甲氧基苄基保护,得到中间体19。
其中,糖基化条件为含卤素氧化剂、路易斯酸(或质子酸)、溶剂、温度的组合。其中,卤素氧化剂为:N-碘代琥珀酰亚胺、N-溴代琥珀酰亚胺、乙酸碘苯等。路易斯酸为:三甲基硅基三氟甲磺酸酯、叔丁基二甲基硅基三氟甲磺酸酯、三乙基硅基三氟甲磺酸酯、三氟化硼乙醚、三氟甲磺酸银等。质子酸为:三氟甲磺酸、三氟乙酸等。溶剂为:乙醚、二氯甲烷、1,4-二氧六环、四氢呋喃或其二元混合物、三元混合物。温度为:-40℃~0℃。
本发明中,如式(IX)所示,化合物19和化合物17,在糖基化条件下得到四糖中间体20。
其中,糖基化条件为含卤素氧化剂、路易斯酸(或质子酸)、溶剂、温度的组合。其中,卤素氧化剂为:N-碘代琥珀酰亚胺、N-溴代琥珀酰亚胺、乙酸碘苯等。路易斯酸为:三甲基硅基三氟甲磺酸酯、叔丁基二甲基硅基三氟甲磺酸酯、三乙基硅基三氟甲磺酸酯、三氟化硼乙醚、三氟甲磺酸银等。质子酸为:三氟甲磺酸、三氟乙酸等。溶剂为:乙醚、二氯甲烷、1,4-二氧六环、四氢呋喃或其二元混合物、三元混合物。温度为:-20℃~0℃。
本发明中,如式(X)所示,化合物20经过碱处理得到四醇21;经硫酸化试剂处理,得到硫酸化中间体22;再在催化剂条件下氢解脱苄,得到化学结构式(I)或/和化学结构简式(II)表示的硫酸化岩藻-半乳四糖。
其中,碱为甲醇钠、乙醇钠、碳酸钠、碳酸钾等。其中,硫酸化试剂为:三氧化硫-吡啶络合物、三氧化硫-三甲胺络合物、三氧化硫-三乙胺络合物等。其中,催化剂为:钯/炭、氢氧化钯/炭、钯黑、铂/炭等。
本发明还提供了式(I)硫酸化岩藻-半乳四糖在制备抑制HSV-1感染的药物中的应用。本发明式(I)表示的化合物抑制HSV-1感染的活性进行了测试,结果表明式(I)化合物对HSV-1感染非洲绿猴肾细胞有明显的活性抑制效果,其抑制HSV-1感染的IC50为42μg ml-1,可见本发明硫酸化岩藻-半乳四糖具有抗HSV-1感染的功能,具有作为治疗HSV-1潜在药物的应用前景。
具体实施方式
结合以下具体实施例,对本发明作进一步的详细说明,本发明的保护内容不局限于以下实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。实施本发明的过程、条件、试剂、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
实施例1对甲苯基2,3,4-三-O-乙酰基-1-硫-β-L-吡喃岩藻糖苷(2)的制备。
0℃,将L-岩藻糖(1.4g,7.73mmol)和DMAP(134mg,1.10mmol)溶解在吡啶(30mL)中,并加入Ac2O(15mL).将该溶液升至室温,并搅拌12h,然后浓缩至干.残余物用EtOAc溶解,用5%柠檬酸水溶液洗涤,再用饱和NaHCO3水溶液和食盐水洗涤.有机层用硫酸镁干燥,过滤,浓缩得到粗产品1(2.62g).0℃,BF3·OEt2(1.96mL,15.47mmol)滴加到粗产品1和甲苯硫酚(1.15g,9.28mmol)溶解于CH2Cl2(30mL)的溶液中。在0℃下搅拌2小时后,溶液升至室温,继续搅拌12小时,并用NaHCO3(sat.aq.)终止反应.有机相用5%NaOH水溶液饱和食盐水洗涤,再用硫酸镁干燥,过滤,蒸干.用i-PrOH和正己烷重结晶,得到2(2.49g,81%,2步)。1H NMR(400MHz,CDCl3)δ7.44-7.38(m,2H),7.13(d,J=7.9Hz,2H),5.25(dd,J=3.3,0.9Hz,1H),5.20(t,J=9.9Hz,1H),5.04(dd,J=9.9,3.4Hz,1H),4.63(d,J=9.9Hz,1H),3.80(qd,J=6.3,0.8Hz,1H),2.34(s,3H),2.14(s,3H),2.09(s,3H),1.97(s,3H),1.23(d,J=6.4Hz,3H).HRMS(ESI-TOF):m/z Calcd for C19H24O7NaS[M+Na]+:419.1140,Found:419.1126.
实施例2对甲苯基2-O-苄基-3,4-O-亚异丙基-1-硫-β-L-吡喃岩藻糖苷(3)的制备。
化合物2(1.18g,2.97mmol)溶解在甲醇(20mL)中,并加入MeONa(16mg,0.30mmol)。搅拌12小时,用Dowex50WX8酸性树脂中和,过滤,蒸干.残余物溶解在丙酮(20mL)中,并加入2,2-二甲氧基丙烷(0.73mL,5.932mmol)和对甲苯磺酸(56mg,0.30mmol)。室温搅拌4小时,加入三乙胺(5mL)中和,蒸干.残余物溶解在DMF(10mL)中,0℃,加入NaH(60%,237mg,5.93mmol)。30min后,加入BnBr(0.71mL,5.93mmol),并在室温下搅拌5小时.混合物缓慢加入到水中,并用乙酸乙酯萃取.有机相用硫酸镁干燥,过滤,蒸干。用正己烷重结晶得到3(1.12g,95%)。1H NMR(400MHz,CDCl3)δ7.47-7.40(m,4H),7.36-7.26(m,3H),7.09(d,J=7.9Hz,2H),4.82and4.67(ABq,J=11.3Hz,2H),4.52(d,J=9.7Hz,1H),4.21(dd,J=5.9,5.9Hz,1H),4.03(dd,J=5.6,2.1Hz,1H),3.79(qd,J=6.5,2.1Hz,1H),3.48(dd,J=9.7,6.5Hz,1H),2.32(s,3H),1.41(s,3H),1.39(d,J=6.6Hz,3H),1.35(s,3H).HRMS(ESI-TOF):m/z Calcd for C23H28O4NaS[M+Na]+:423.1606,Found:423.1599.
实施例3对甲苯基2-O-苄基-1-硫-β-L-吡喃岩藻糖苷(4)的制备
化合物3(1.11g,2.77mmol)溶解在(25mL)中,并加入对甲苯磺酸(53mg,0.28mmol).在室温下搅拌24小时,用三乙胺(5mL)中和,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶2∶1)得到4(895mg,90%)。1H NMR(400MHz,CDCl3)δ7.47(d,J=8.1Hz,2H),7.42-7.27(m,5H,ArH),7.12(d,J=7.9Hz,2H),4.96and4.69(ABq,J=11.0Hz,2H),4.53(d,J=9.6Hz,1H),3.72(dd,J=5.1,3.4Hz,1H),3.66-3.56(m,2H),3.50(t,J=9.3Hz,1H),2.48(d,J=5.3Hz,1H),2.34(s,3H),2.11(d,J=5.3Hz,1H),1.34(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C20H24O4NaS[M+Na]+:383.1293,Found:383.1294.
实施例4对甲苯基2,3-di-O-对甲苯硫基-1-硫-β-L-吡喃岩藻糖苷(5)的制备。
化合物4(884mg,2.45mmol)和Bu2SnO(763mg,3.07mmol)溶解在甲苯(40mL)中,并加热至130℃,搅拌4小时。冷至60℃,依次加入DMF(15mL),BnBr(0.44mL,3.68mmol),CsF(745mg,4.90mmol)和TBAI(272mg,0.74mmol),并继续在60℃加热12小时。用水终止反应,并用乙酸乙酯萃取。有机相用饱和食盐水洗涤,硫酸镁干燥,过滤,蒸干.硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶5∶1)得到5(1.09g,98%)。1H NMR(400MHz,CDCl3)δ7.49-7.45(m,2H),7.44-7.39(m,2H),7.37-7.26(m,8H),7.10(d,J=7.9Hz,2H),4.84and4.74(ABq,J=10.3Hz,2H),4.71and4.68(ABq,J=11.6Hz,2H),4.53(d,J=9.6Hz,1H),3.81(ddd,J=3.4,3.4,0.9Hz,1H),3.65(dd,J=9.3,9.3Hz,1H),3.58-3.51(m,2H),2.32(s,3H),2.23(dd,J=3.4,0.7Hz,1H),1.36(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C27H30O4NaS[M+Na]+:473.1763,Found:473.1777.
实施例5对甲苯基4-O-苯甲酰基-2,3-二-O-苄基-1-硫-β-L-吡喃岩藻糖苷(6)的制备。
苯甲酰氯(0.83mL,7.20mmol)慢慢滴加到5(1.08g,2.40mmol)和吡啶(1.4mL,16.79mmol)以及DMAP(59mg,0.48mmol)溶解在二氯甲烷(20mL)的溶液中。在室温下搅拌18h,用NaHCO3终止反应。有机相依次用5%柠檬酸溶液,NaHCO3饱和溶液和饱和食盐水洗涤,MgSO4干燥,过滤,蒸干。用甲醇重结晶,得到6(1.1g,83%)。1H NMR(400MHz,CDCl3)δ8.06-8.01(m,2H),7.63-7.54(m,3H),7.48-7.43(m,2H),7.42-7.37(m,2H),7.36-7.24(m,5H),7.24-7.19(m,3H),7.15(d,J=7.9Hz,2H),5.61(dd,J=3.2,0.7Hz,1H),4.79and4.52(ABq,J=11.3Hz,2H),4.74and4.72(ABq,J=10.4Hz,2H),4.60(d,J=9.4Hz,1H),3.81(qd,J=6.4,0.8Hz,1H),3.75(dd,J=9.1,3.2Hz,1H),3.67(t,J=9.3Hz,1H),2.38(s,3H),1.30(d,J=6.4Hz,3H).HRMS(ESI-TOF):m/zCalcd for C34H34O5NaS[M+Na]+:577.2025,Found:577.2019.
实施例64-O-苯甲酰基-2,3-二-O-苄基-α/β-L-吡喃岩藻糖苷(7)的制备。
方法一:0℃,向6(208mg,0.38mmol)溶解在丙酮/水(9∶1,5mL)的溶液中加入NBS(133mg,0.75mmol)。0℃搅拌5小时,溶液倾入CH2Cl2中。有机相一次用NaHCO3饱和溶液,Na2S2O3饱和溶液,饱和食盐水洗涤,MgSO4干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶3∶1)得到7α和7β(5/3)的混合物(146mg,87%)。HRMS(ESI-TOF):m/z Calcd for C27H28O6Na[M+Na]+:471.1784,Found:471.1805.7α:1H NMR(400MHz,CDCl3)δ8.09-8.04(m,2H),7.59-7.56(m,1H),7.49-7.45(m,2H),7.34-7.24(m,10H),5.64(dd,J=3.2,1.0Hz,1H),5.32(d,J=3.6Hz,1H),4.85and4.70(ABq,J=11.6Hz,2H),4.83and4.59(ABq,J=11.5Hz,2H),4.38(qd,J=6.6,0.5Hz,1H),4.04(dd,J=9.8,3.3Hz,1H),3.90(dd,J=9.8,3.6Hz,1H),2.96(s,1H),1.20(d,J=6.5Hz,3H).7β:1H NMR(400MHz,CDCl3)δ8.15-8.10(m,2H),7.61-7.58(m,1H),7.46-7.42(m,2H),7.28-7.22(m,10H),5.58(dd,J=3.3,1.0Hz,1H),4.90-4.79(m,3H),4.74(dd,J=7.4,3.0Hz,1H),4.57(B ofABq,J=11.6Hz,1H),3.81(qd,J=6.3,0.8Hz,1H),3.71(dd,J=9.6,3.4Hz,1H),3.63(dd,J=9.6,7.4Hz,1H),3.16(d,J=4.5Hz,1H),1.27(d,J=6.4Hz,3H).
方法二:0℃,向6(208mg,0.38mmol)溶解在二氯甲烷/水(9∶1,5mL)的溶液中加入NIS(169mg,0.75mmol)和三氟甲磺酸银(10.3mg,0.04mmol)。0℃搅拌2小时,溶液倾入CH2Cl2中。有机相一次用NaHCO3饱和溶液,Na2S2O3饱和溶液,饱和食盐水洗涤,MgSO4干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶3∶1)得到7α和7β(5/3)的混合物(159mg,95%)。
实施例74-O-苯甲酰基-2,3-二-O-苄基-α-L-吡喃岩藻糖基三氯乙酰亚胺酸酯(8)的制备。
方法一:化合物7(140mg,0.31mmol)溶解在干燥的CH2Cl2(5mL)中。向此溶液中加入Cl3CCN(0.26mL,3.0mmol)和1,8-二氮杂二环十一碳-7-烯(15μL,0.11mmol)。室温搅拌5小时,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷1∶20)得到8(138.4mg,75%)。1H NMR(400MHz,CDCl3)δ8.58(s,1H),8.09-8.02(m,2H),7.62-7.57(m,1H),7.50-7.43(m,2H),7.34-7.27(m,7H),7.25-7.20(m,3H),6.55(d,J=3.3Hz,1H),5.69(dd,J=3.0,1.1Hz,1H),4.82and4.63(ABq,J=11.7Hz,2H),4.78and4.74(ABq,J=11.9Hz,2H),4.34(qd,J=6.5,0.7Hz,1H),4.15(dd,J=10.0,3.1Hz,1H),4.09(dd,J=10.0,3.3Hz,1H),1.22(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C29H28NO6NaCl3[M+Na]+:614.0880,Found:614.0880.
方法二:化合物7(140mg,0.31mmol)溶解在干燥的CH2Cl2(5mL)中。向此溶液中加入Cl3CCN(0.26mL,3.0mmol)和氢化钠(2.5mg,0.11mmol)。室温搅拌3小时,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷1∶20)得到8(121mg,66%)。
实施例8N-苯基-O-(4-O-苯甲酰基-2,3-二-O-苄基-α/β-L-吡喃岩藻糖基)三氟乙酰亚胺酸酯(9)的制备。
方法一:向化合物7(331mg,0.74mmol)的CH2Cl2(10mL)溶液中加入N-苯基三氟乙酰亚胺酰氯(184mg,0.89mmol)和Cs2CO3(481mg,1.48mmol)。室温搅拌2小时,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷1∶15)得到9β和9α(4.3/1.0)的混合物(396mg,87%)。9β:1H NMR(400MHz,CDCl3)δ8.13(d,J=7.7Hz,2H),7.64-7.58(m,1H),7.49(t,J=7.7Hz,2H),7.35-7.22(m,12H),7.14-7.07(m,1H),6.83(d,J=7.6Hz,2H),5.67(brs,1H),5.58(s,1H),4.85-4.78(m,3H),4.57(d,J=11.6Hz,1H),3.90(t,J=8.3Hz,1H),3.82-3.55(m,2H,H-5),1.26(d,J=6.3Hz,3H).HRMS(ESI-TOF):m/zCalcd for C35H32F3NO6Na[M+Na]+:642.2079,Found:642.2093.9α:1H NMR(400MHz,CDCl3)δ8.04(d,J=7.7Hz,2H),7.58(t,J=7.4Hz,1H),7.45(t,J=7.7Hz,2H),7.35-7.21(m,12H),7.09(t,J=7.4Hz,1H),6.77(d,J=6.6Hz,2H),6.53(brs,1H),5.68(s,1H),4.83and4.63(ABq,J=11.4Hz,2H),4.82and4.73(ABq,J=11.9Hz,2H),4.27(brs,1H),4.11(d,J=9.6Hz,1H),4.06(d,J=9.6Hz,1H),1.22(d,J=6.3Hz,3H).13C HRMS(ESI-TOF):m/z Calcd for C35H32F3NO6Na[M+Na]+:642.2079,Found:642.2098.
方法二:向化合物7(331mg,0.74mmol)的CH2Cl2(10mL)溶液中加入N-苯基三氟乙酰亚胺酰氯(184mg,0.89mmol)和K2CO3(xx mg,1.48mmol)。室温搅拌2小时,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷1:15)得到9β(387mg,85%)。
实施例9对甲苯基4-O-苯甲酰基-2-O-苄基-1-硫-β-L-吡喃岩藻糖苷(10)的制备
如化合物3的合成所述,化合物2(337mg,0.85mmol)的脱乙酰化可以得到粗产物三醇。将该三醇与甲苯共沸,然后用乙腈(10mL)溶解。向该溶液中加入原苯甲酸三甲酯(0.29mL,1.7mmol)和DL-樟脑磺酸(20mg,0.085mmol),室温下搅拌1小时。然后加入三乙胺(5mL),并旋蒸除去溶剂。将所得的醇溶于DMF(8mL),然后在0℃下分批加入NaH(60%,68mg,1.7mmol)。30分钟后,加入BnBr(0.2mL,1.7mmol),反应液在室温下搅拌12小时。反应完成后,向该反应液中加入1N HCl(2.6mL),搅拌一小时,然后加入EtOAc和H2O,分液,有机相用无水MgSO4干燥,过滤,浓缩。粗产物经硅胶柱层析(乙酸乙酯/正己烷/二氯甲烷1∶5∶1)后得到纯净的油状物10(390mg,82%)。1H NMR(400MHz,CDCl3)δ8.06-8.00(m,2H),7.65-7.54(m,3H),7.50-7.43(m,2H),7.39-7.26(m,5H),7.19-7.14(m,2H),5.42(dd,J=3.4,0.8Hz,1H),4.95and4.66(ABq,J=10.8Hz,2H),4.61(d,J=9.6Hz,1H),3.93(td,J=9.1,3.6Hz,1H),3.84(qd,J=6.4,0.9Hz,1H),3.63(t,J=9.3Hz,1H),2.39(s,3H),2.29(d,J=3.7Hz,1H),1.28(d,J=6.4Hz,3H).HRMS(ESI-TOF):m/z Calcd for C27H28O5NaS[M+Na]+:487.1555,Found:487.1578.
实施例10p-甲氧基苯基2,3,4-三-O-乙酰基-α-L-吡喃岩藻糖苷(11)的制备
在氮气保护和0℃的条件下,向1(417mg,1.25mmol)和对甲氧基苯酚(234mg,1.88mmol)的CH2Cl2(10mL)溶液中逐滴加入BF3·OEt2(0.32mL,2.51mmol)。在0℃下搅拌2小时后,将反应液自然升温至室温,在室温下继续搅拌12小时,然后用NaHCO3(sat.aq.)淬灭。将有机相分离,依次用5%NaOH(aq)和饱和食盐水洗涤,然后用无水MgSO4干燥,过滤,旋蒸。所得的粗产物经硅胶柱层析(乙酸乙酯/正己烷/二氯甲烷1∶9∶1)后得到纯的化合物11(386mg,78%)。1H NMR(400MHz,CDCl3)δ7.01-6.93(m,2H),6.86-6.78(m,2H),5.62(d,J=3.6Hz,1H),5.57(dd,J=10.9,3.4Hz,1H),5.37(dd,J=3.3,1.1Hz,1H),5.26(dd,J=10.9,3.7Hz,1H),4.32(qd,J=6.5,0.7Hz,1H),3.77(s,3H),2.19(s,3H),2.07(s,3H),2.02(s,3H),1.14(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C19H24O9Na[M+Na]+:419.1318,Found:419.1308。
实施例11p-甲氧基苯基4-O-苯甲酰基-2-O-苄基-α-L-吡喃岩藻糖苷(12)的制备
参照化合物10的合成方法,从化合物11(350mg,0.88mmol)出发,得到了化合物12(298mg,70%)。1H NMR(400MHz,CDCl3)δ8.10-8.04(m,2H),7.62-7.56(m,1H),7.50-7.42(m,2H),7.36-7.26(m,5H),7.06-7.00(m,2H),6.87-6.81(m,2H),5.55(dd,J=3.4,1.1Hz,1H),5.50(d,J=3.5Hz,1H),4.71(s,2H),4.49(dd,J=10.0,3.5Hz,1H),4.32(qd,J=6.6,0.8Hz,1H),3.96(dd,J=10.1,3.5Hz,1H),3.79(s,3H),2.43(br.s,1H),1.17(d,J=6.6Hz,3H).HRMS(ESI-TOF):m/z Calcd for C27H28O7Na[M+Na]+:487.1733,Found:487.1728.
实施例12p-甲苯基3,6-二-O-苄基-2-O-(4-甲氧基苄基)-1-硫-β-D-吡喃半乳糖苷(15)的制备
方法一:将化合物13(300mg,0.51mmol)溶解在甲醇(5mL)中,并加入对甲苯磺酸(10mg,0.05mmol).室温搅拌36小时,用三乙胺(2mL)中和,蒸干,得到粗产品14。1H NMR(400MHz,CDCl3)δ7.49-7.43(m,2H),7.40-7.26(m,7H),7.10(d,J=8.0Hz,2H),6.90-6.84(m,2H),4.78and4.68(ABq,J=9.9Hz,2H),4.71(s,2H),4.57(d,J=9.7Hz,1H),4.05-4.00(m,1H),3.99-3.92(m,1H),3.810(s,3H3),3.80-3.73(m,1H),3.70(t,J=9.3Hz,1H),3.56(dd,J=8.9,3.3Hz,1H),3.45(dd,J=6.5,4.5Hz,1H),2.57(s,1H),2.32(s,3H),2.10(dd,J=8.6,3.8Hz,1H).HRMS(ESI-TOF):m/z Calcd for C28H32O6NaS[M+Na]+:519.1817,Found:519.1837.
该粗产品14溶解在CH2Cl2(10mL)中,并加入BnBr(122μL,1.03mmol)和四丁基硫酸氢铵(35mg,0.10mmol)和5%NaOH水溶液(5mL)。50℃搅拌30h后,用CH2Cl2和H2O稀释。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。用甲醇重结晶得到化合物15(194mg,61%,2步)。1H NMR(400MHz,CDCl3)δ7.50-7.44(m,2H),7.37-7.27(m,12H),7.05(d,J=7.9Hz,2H),6.89-6.84(m,2H),4.77(d,J=9.9Hz,1H),4.74-4.65(m,3H),4.59-4.52(m,3H,H-1),4.09-4.06(m,1H4),3.80(s,3H),3.83-3.73(m,2H),3.69(t,J=9.3Hz,1H),3.58-3.51(m,2H),2.48(dd,J=2.5,0.6Hz,1H),2.30(s,3H).HRMS(ESI-TOF):m/z Calcd for C35H38O6NaS[M+Na]+:609.2287,Found:609.2280.
方法二:14的制备同上。该粗产品14溶解在CH2Cl2(10mL)中,并加入BnBr(122μL,1.03mmol)和四丁基溴化铵(32mg,0.10mmol)和5%KOH水溶液(5mL)。50℃搅拌30h后,用CH2Cl2和H2O稀释。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。用甲醇重结晶得到化合物15(210mg,66%,2步)。
方法三:14的制备同上。该粗产品14溶解在CH2Cl2(10mL)中,并加入BnBr(122μL,1.03mmol)和四乙基氯化铵(166mg,0.10mmol)和10%Na2CO3水溶液(5mL)。50℃搅拌30h后,用CH2Cl2和H2O稀释。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。用甲醇重结晶得到化合物15(229mg,72%,2步)。
方法四:14的制备同上。该粗产品14溶解在CH2Cl2(10mL)中,并加入BnBr(122μL,1.03mmol)和十二烷基三甲基溴化铵(31mg,0.10mmol)和15%K2CO3水溶液(5mL)。50℃搅拌30h后,用CH2Cl2和H2O稀释。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。用甲醇重结晶得到化合物15(146mg,46%,2步)。
实施例13p-甲苯基4-O-乙酰基-3,6-二-O-苄基-2-O-(4-甲氧基苄基)-1-硫-β-D-吡喃半乳糖苷(16)的制备
将化合物15(137mg,0.23mmol),三乙胺(0.39mL,2.80mmol)以及DMAP(3mg,0.023mmol)溶解在CH2Cl2(5mL)中,并加入Ac2O(0.11mL,1.17mmol)。室温搅拌5小时,然后用NaHCO3饱和溶液淬灭。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶10∶1)得到化合物16(172mg,88%)。1H NMR(400MHz,CDCl3)δ7.50-7.44(m,2H),7.37-7.26(m,12H),7.05(d,J=7.9Hz,2H),6.91-6.83(m,2H),5.61(d,J=1.7Hz,1H),4.77and4.49(ABq,J=11.0Hz,2H),4.70and4.65(ABq,J=9.8Hz,2H),4.64-4.57(m,1H,H-1),4.54and4.45(ABq,J=11.7Hz,2H),3.80(s,3H),3.71(dd,J=6.3,6.3Hz,1H),3.64-3.57(m,3H),3.51(dd,J=9.6,6.7Hz,1H),2.30(s,3H),2.08(s,3H).HRMS(ESI-TOF):m/z Calcd for C37H40O7NaS[M+Na]+:651.2392,Found:651.2381.
实施例14p-甲苯基(4-O-苯甲酰基-2,3-二-O-苄基-α-L-吡喃岩藻糖基)-(1→3)-4-O-苯甲酰基-2-O-苄基-1-硫-β-L-吡喃岩藻糖苷(17)的制备
方法一:将化合物8(133mg,0.224mmol)和化合物10(87mg,0.187mmol)溶解在CH2Cl2(5mL)中,并加入 分子筛(500mg),室温搅拌1小时。将混合物冷至-78℃,并加入TMSOTf(3.4μL,18.7μmol)。在-78℃下搅拌1小时,并在30分钟内升温至0℃。用NaHCO3饱和溶液淬灭,用CH2Cl2稀释,过滤。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶10∶2)得到化合物17(112mg,67%)。1H NMR(400MHz,CDCl3)δ8.05-7.98(m,2H),7.96-7.87(m,2H),7.66-7.59(m,2H),7.59-7.49(m,2H),7.45-7.28(m,9H),7.22-7.06(m,12H),5.67(d,J=2.9Hz,1H),5.28(d,J=3.4Hz,1H),5.16(dd,J=3.0,0.9Hz,1H),5.02and4.55(ABq,J=10.5Hz,2H),4.65(d,J=9.5Hz,1H),4.63and4.41(ABq,J=11.1Hz,2H),4.52and4.43(ABq,J=12.1Hz,2H),4.12(qd,J=6.4,0.8Hz,1H),4.01(dd,J=9.5,3.2Hz,1H),3.90(dd,J=10.1,3.2Hz,1H),3.86-3.77(m,3H),2.41(s,3H),1.28(d,J=6.4Hz,3H),0.91(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C54H54O10NaS[M+Na]+:917.3335,Found:917.3351.
方法二:将化合物8(133mg,0.224mmol)和化合物10(87mg,0.187mmol)溶解在二氯甲烷/1,4-二氧六环(1∶1,5mL)中,并加入 分子筛(500mg),室温搅拌1小时。将混合物冷至-60℃,并加入TBSOTf(3.4μL,18.7μmol)。在-60℃下搅拌1小时,并在30分钟内升温至0℃。用NaHCO3饱和溶液淬灭,用CH2Cl2稀释,过滤。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶10∶2)得到化合物17(94mg,56%)。
方法三:将化合物8’(84mg,0.135mmol)和化合物10(52mg,0.112mmol)溶解在二氯甲烷/乙醚/1,4-二氧六环(1∶1∶1,3mL)中,并加入 分子筛(400mg),室温搅拌1小时。将混合物冷至-40℃,并加入三氟甲磺酸银(2.8mg,11.2μmol)。在-40℃下搅拌2.5小时,并在30分钟内升温至0℃。用NaHCO3饱和溶液淬灭,用CH2Cl2稀释,过滤。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶10∶2)得到化合物17(131mg,78%)。
方法四:将化合物8’(84mg,0.135mmol)和化合物10(52mg,0.112mmol)溶解在CH2Cl2/四氢呋喃(1∶1,3mL)中,并加入 分子筛(400mg),室温搅拌1小时。将混合物冷至-20℃,并加入三氟甲磺酸(xxxμL,11.2μmol)。在-20℃下搅拌2.5小时,并在30分钟内升温至0℃。用NaHCO3饱和溶液淬灭,用CH2Cl2稀释,过滤。有机相用饱和食盐水洗涤,干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶10∶2)得到化合物17(94mg,56%)。
实施例15p-甲氧基苯基[4-O-乙酰基-3,6-二-O-苄基-2-O-(4-甲氧基苄基)-α-D-吡喃半乳糖基]-(1→3)-4-O-苯甲酰基-2-O-苄基-α-L-吡喃岩藻糖苷(18)的制备
方法一:将化合物16(159mg,0.253mmol)和化合物12(98mg,0.211mmol)溶解在CH2Cl2(5mL)中,并加入 分子筛(500mg),并在室温搅拌0.5小时。反应混合物冷至-20℃,并依次加入NIS(57mg,0.253mmol)和三氟甲磺酸银(5mg,21.1μmol)。在-20℃下搅拌1小时,用NaHCO3/Na2S2O3饱和溶液终止反应,EtOAc稀释,过滤。有机相饱和食盐水洗涤,干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶5∶1)得到化合物18(162mg,79%)。1H NMR(400MHz,CDCl3)δ8.09-8.03(m,2H),7.63-7.57(m,1H),7.49-7.42(m,2H),7.37-7.32(m,2H),7.30-7.19(m,13H),7.01-6.95(m,4H),6.85-6.79(m,2H),6.68-6.62(m,2H),5.60(dd,J=2.8,1.1Hz,1H),5.50(dd,J=3.4,0.8Hz,1H),5.46(d,J=3.2Hz,1H),5.34(d,J=3.6Hz,1H),4.64-4.45(m,9H),4.28(d,J=11.1Hz,1H),4.12(qd,J=6.6,0.7Hz,1H),4.05(dd,J=10.1,3.6Hz,1H),3.78(s,3H),3.77-3.73(m,2H),3.72(s,3H),3.62-3.57(m,2H),2.03(s,3H),1.02(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C57H60O14Na[M+Na]+:991.3881,Found:991.3931.
方法二:将化合物16(159mg,0.253mmol)和化合物12(98mg,0.211mmol)溶解在CH2Cl2/乙醚/1,4-二氧六环(3∶1∶1,5mL)中,并加入 分子筛(500mg),并在室温搅拌0.5小时。反应混合物冷至-40℃,并依次加入NBS(45mg,0.253mmol)和TESOTf(5.5mg,21.1μmol)。在-20℃下搅拌1小时,用NaHCO3/Na2S2O3饱和溶液终止反应,EtOAc稀释,过滤。有机相饱和食盐水洗涤,干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶5∶1)得到化合物18(159mg,76%)。
方法三:将化合物16(159mg,0.253mmol)和化合物12(98mg,0.211mmol)溶解在CH2Cl2/四氢呋喃(1∶1,5mL)中,并加入 分子筛(500mg),并在室温搅拌0.5小时。反应混合物冷至0℃,并依次加入NIS(65mg,0.253mmol)和三氟乙酸(2.4mg,21.1μmol)。在0℃下搅拌1小时,用NaHCO3/Na2S2O3饱和溶液终止反应,EtOAc稀释,过滤。有机相饱和食盐水洗涤,干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶5∶1)得到化合物18(140mg,68%)。
实施例16p-甲氧基苯基(4-O-乙酰基-3,6-二-O-苄基-α-D-吡喃半乳糖基)-(1→3)-4-O-苯甲酰基-2-O-苄基-α-L-吡喃岩藻糖苷(19)的制备
0℃,DDQ(41g,0.19mmol)加入到化合物18(157mg,0.16mmol)溶解在混合溶剂(CH2Cl2/pH7.0磷酸盐缓冲溶液=20/1,5mL)的溶液中。0℃搅拌2小时,用NaHCO3饱和溶液和CH2Cl2稀释。有机相依次用NaHCO3饱和溶液和饱和食盐水洗涤,MgSO4干燥,过滤,蒸干。硅胶柱层析纯化(乙酸乙酯/正己烷/二氯甲烷1∶5∶1)得到化合物19(103mg,75%)。1H NMR(400MHz,CDCl3)δ8.06-8.01(m,2H),7.65-7.57(m,1H),7.50-7.43(m,2H),7.36-7.20(m,15H),7.03-6.96(m,2H,ArH),6.87-6.79(m,2H),5.57(d,J=2.4Hz,1H),5.53(d,J=2.7Hz,1H),5.43(d,J=3.5Hz,1H),5.39(d,J=3.9Hz,1H),4.71and4.61(ABq,J=11.6Hz,2H),4.66and4.26(ABq,J=11.3Hz,2H,),4.59and4.50(ABq,J=11.8Hz,2H),4.52(dd,J=10.1,3.5Hz,1H),4.40(t,J=6.2Hz,1H),4.18(q,J=6.3Hz,1H),4.04(dd,J=10.2,3.5Hz,1H),3.89(ddd,J=10.0,8.1,3.9Hz,1H),3.79(s,3H),3.62-3.51(m,2H),3.48(dd,J=10.0,3.2Hz,1H),2.14(d,J=8.1Hz,1H),2.04(s,3H),1.07(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C49H52O13Na[M+Na]+:871.3306,Found:871.3295.
实施例17p-甲氧基苯基(4-O-苯甲酰基-2,3-二-O-苄基α-L-吡喃岩藻糖基)-(1→3)-(4-O-苯甲酰基-2-O-苄基-α-L-吡喃岩藻糖基)-(1→2)-(4-O-乙酰基-3,6-二-O-苄基-α-D-吡喃半乳糖基)-(1→3)-4-O-苯甲酰基-2-O-苄基-α-L-吡喃岩藻糖苷(20)的制备
方法一:将化合物17(65.1mg,72.7μmol)和化合物19(55mg,64.8μmol)溶解在无水溶剂中(二氯甲烷/乙醚=1/1,4mL),并加入 分子筛(400mg),在室温下搅拌1小时。溶液冷至-10℃,并加入NIS(18mg,78.8μmol)和三氟甲磺酸银(2mg,6.5μmol)。在-10℃下搅拌3小时,用三乙胺中和,硅藻土过滤。硅胶柱纯化(乙酸乙酯/正己烷/二氯甲烷1∶10∶1)得到化合物20(82mg,78%)。1H NMR(400MHz,CDCl3)δ8.11-8.05(m,2H),8.00-7.93(m,2H),7.91-7.84(m,2H),7.66-7.60(m,1H),7.59-7.52(m,1H),7.51-7.39(m,7H),7.39-7.19(m,10H),7.18-7.07(m,14H),7.07-7.01(m,2H),7.01-6.92(m,4H),6.87(d,J=7.2Hz,2H),6.82-6.75(m,2H),5.66(d,J=2.4Hz,1H),5.57(d,J=2.7Hz,1H),5.46(d,J=3.5Hz,1H),5.44(d,J=3.6Hz,1H),5.40(d,J=3.7Hz,1H),5.18(d,J=3.0Hz),5.12-5.08(m,2H),4.81and4.77(ABq,J=12.5Hz,2H),4.71(d,J=11.4Hz,1H),4.68-4.59(m,3H),4.53and4.38(ABq,J=12.2Hz,2H),4.52(d,J=11.7Hz,1H),4.47(d,J=11.5Hz,1H),4.36(d,J=12.0Hz,1H),4.33-4.10(m,8H),3.94(dd,J=10.3,3.2Hz,1H),3.88-3.73(m,7H),3.66-3.60(m,2H),2.08(s,3H),1.03(d,J=6.5Hz,3H),0.94(d,J=6.5Hz,3H),0.72(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C96H98O23Na[M+Na]+:1641.6397,Found:1641.6345.
方法二:将化合物17(65.1mg,72.7μmol)和化合物19(55mg,64.8μmol)溶解在无水溶剂CH2Cl2/乙醚/1,4-二氧六环(3∶1∶1,5mL),并加入 分子筛(400mg),在室温下搅拌1小时。溶液冷至0℃,并加入NBS(14mg,78.8μmol)和TBSOTf(1.8mg,6.5μmol)。在0℃下搅拌3小时,用三乙胺中和,硅藻土过滤。硅胶柱纯化(乙酸乙酯/正己烷/二氯甲烷1∶10∶1)得到化合物20(85mg,81%)。
方法三:将化合物17(65.1mg,72.7μmol)和化合物19(55mg,64.8μmol)溶解在无水溶剂CH2Cl2/四氢呋喃(1∶1,5mL),并加入 分子筛(400mg),在室温下搅拌1小时。溶液冷至-20℃,并加入NIS(18mg,78.8μmol)和三氟甲磺酸(0.95mg,6.5μmol)。在-20℃下搅拌3小时,用三乙胺中和,硅藻土过滤。硅胶柱纯化(乙酸乙酯/正己烷/二氯甲烷1∶10∶1)得到化合物20(76mg,73%)。
实施例18p-甲氧基苯基(2,3-二-O-苄基-α-L-吡喃岩藻糖基)-(1→3)-(2-O-苄基-α-L-吡喃岩藻糖基)-(1→2)-(3,6-二-O-苄基-α-D-吡喃半乳糖基)-(1→3)-2-O-苄基-α-L-吡喃岩藻糖苷(21)的制备
方法一:化合物20(74mg,45.4μmol)溶解在甲醇(5mL)中,并加入MeONa(12mg,230μmol).反应液50℃下搅拌3天。加入Dowex50WX8酸性树脂中和,过滤,蒸干。硅胶柱层析纯化(甲醇/二氯甲烷1∶100)得到四醇21(47mg,82%)。1H NMR(400MHz,CDCl3)δ7.39-7.10(m,30H),7.01-6.95(m,2H),6.85-6.79(m,2H),5.37(d,J=3.7Hz,1H),5.26(d,J=3.3Hz,1H),5.02(d,J=3.6Hz,1H),4.92and4.84(ABq,J=12.8Hz,2H),4.76and4.62(ABq,J=11.8Hz,2H),4.74-4.67(m,2H),4.68(d,J=11.7Hz,1H),4.63(d,J=11.6Hz,1H),4.59-4.54(m,1H),4.54-4.45(m,5H),4.28(qd,J=5.9,0.8Hz,1H),4.14-4.06(m,2H),4.06-4.00(m,2H),4.00-3.88(m,5H),3.88-3.71(m,7H),3.71-3.63(m,2H),3.22(br.s,1H),3.19(d,J=1.7Hz,1H),2.58(s,1H),2.32(s,1H),1.62(br.s,1H),1.36(d,J=6.5Hz,3H),0.94(d,J=6.6Hz,3H),0.81(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C73H84O19Na[M+Na]+:1287.5505,Found:1287.5526.
方法二:化合物20(74mg,45.4μmol)溶解在甲醇(5mL)中,并加入碳酸钾(32mg,230μmol)。反应液50℃下搅拌4天。加入Dowex50WX8酸性树脂中和,过滤,蒸干。硅胶柱层析纯化(甲醇/二氯甲烷1∶100)得到四醇21(45mg,78%)。
实施例19
p-甲氧基苯基(4-O-sodium sulfonato-α-L-吡喃岩藻糖基)-(1→3)-(4-O-sodium sulfonato-α-L-吡喃岩藻糖基)-(1→2)-(4-O-sodium sulfonato-α-D-吡喃半乳糖基)-(1→3)-4-O-benzyl-α-L-吡喃岩藻糖基(式(I)化合物)的制备
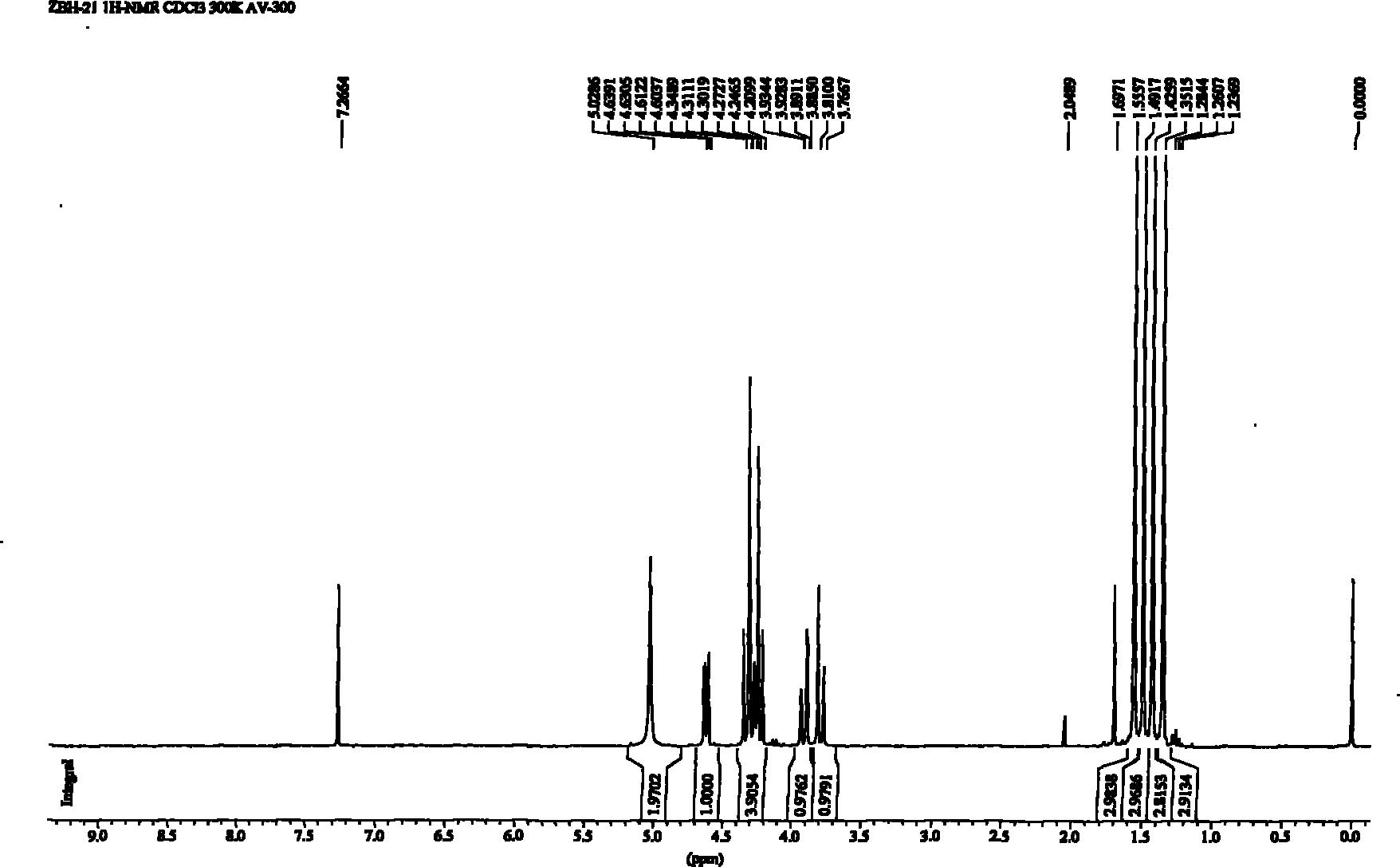
方法一:化合物23(24mg,19.0μmol)和SO3·Me3N络合物(53mg,379μmol)溶解在DMF(1mL)中,并加热至50℃,搅拌3天。0℃,反应用三乙胺和水淬灭,蒸干。残余物用Dowex50WX8(Na+form)树脂处理,蒸干,并溶解在MeOH/H2O(1/2,6mL)。向上述溶液中加入20%Pd(OH)2/C(450mg),置换氢气,搅拌2天,硅藻土过滤,蒸干。残余物用Dowex50WX8(Na+)树脂处理,再用Sephadex G-25凝胶过滤,冷冻干燥得到式(I)化合物(17mg,80%)。1H NMR(400MHz,D2O,300K)δ7.14(d,J=9.1Hz,2H),7.01(d,J=9.0Hz,2H),5.51(d,J=3.8Hz,1H),5.37(d,J=3.7Hz,1H),5.29(d,J=3.9Hz,1H),5.19(d,J=3.9Hz,1H),4.80-4.75(overlapped by HOD,3H),4.64(d,J=2.7Hz,1H),4.52(dd,J=8.5,3.6Hz,1H),4.50-4.36(m,5H),4.18(dd,J=10.5,3.9Hz,1H),4.14(dd,J=10.5,2.7Hz,1H),4.05(dd,J=10.6,3.1Hz,1H),4.00(dd,J=10.5,3.8Hz,1H),3.93(dd,J=10.4,3.9Hz,1H),3.89-3.76(m,6H),1.30(d,J=6.5Hz,6H),1.25(d,J=6.5Hz,3H).HRMS(ESI-TOF):m/z Calcd for C31H44O31Na3S4[M+3Na]-:1109.0443,Found:1109.0490.
方法二:化合物23(24mg,19.0μmol)和三氧化硫吡啶络合物(60.3mg,379μmol)溶解在DMF(1mL)中,并加热至50℃,搅拌3天。0℃,反应用三乙胺和水淬灭,蒸干。残余物用Dowex50WX8(Na+)树脂处理,蒸干,并溶解在MeOH/H2O(1/2,6mL)。向上述溶液中加入10%Pd/C(240mg),置换氢气,搅拌2天,硅藻土过滤,蒸干。残余物用Dowex50WX8(Na+)树脂处理,再用Sephadex G-25凝胶过滤,冷冻干燥得到式(I)化合物(16mg,76%)。
方法三:化合物23(24mg,19.0μmol)和三氧化硫三乙胺络合物(xx mg,379μmol)溶解在DMF(1mL)中,并加热至50℃,搅拌3天。0℃,反应用三乙胺和水淬灭,蒸干。残余物用Dowex50WX8(Na+)树脂处理,蒸干,并溶解在MeOH/H2O(1/2,6mL)。向上述溶液中加入钯黑(26mg),置换氢气,搅拌2天,硅藻土过滤,蒸干。残余物用Dowex50WX8(Na+)树脂处理,再用Sephadex G-25凝胶过滤,冷冻干燥得到式(I)化合物(18mg,83%)。实施例20式(I)化合物抑制HSV-1感染的活性测试
HSV-1和不同浓度的式(I)化合物样品溶液(1.0,5.0,10.0,20.0,40.0和100.0μg ml-1)混合;以商品化的硫酸肝素用作对照样品。得到的混合溶液用于对非洲绿猴肾细胞的感染,感染过程在37℃下进行60分钟。随后,感染的细胞用PBS缓冲溶液冲洗,并用1%琼脂糖进行覆盖。3天之后,统计斑块数目。空白对照所产生的斑块数目定为100%,并作为空白对照。实验结果表明,式(I)化合物对斑块的形成有明显抑制作用,测得式(I)化合物的抑制HSV-1感染的IC50为42μg ml-1。
硫酸化岩藻-半乳四糖及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0