










专利摘要
本发明公开了一种用于氧化脱硫的苯并噻吩类化合物的光解方法。该方法包括苯并噻吩类化合物在氧化剂以及金属酞菁或金属卟啉存在下,经过光照反应达到C-S键断裂的反应。本发明的方法在常温常压的温和条件下,氧化石油产品中最为稳定的一类硫化物-苯并噻吩类化合物,达到苯并噻吩类化合物分子中C-S键氧化断裂,得到极性更大的含硫化合物和不含硫的烃类氧化物,在金属卟啉为催化剂时C-S键断裂转化率达到90%以上,克服了目前苯并噻吩类化合物分子中C-S键氧化断裂反应需要高温、高压和特殊催化剂,对设备的要求高,产率低等问题。
权利要求
1.一种用于氧化脱硫的苯并噻吩类化合物的光解方法,其特征在于,该方法包括苯并噻吩类化合物在氧化剂以及金属酞菁或金属卟啉存在下,经过光照反应达到C-S键断裂的反应。
2.根据权利要求1所述的光解方法,其特征在于,所述的光照反应在10~50℃、常压下进行。
3.根据权利要求1所述的光解方法,其特征在于,所述的光的波长为200~600nm。
4.根据权利要求1所述的光解方法,其特征在于,所述光照反应的反应溶剂为C1-3醇化合物、C6-20烃化合物或二者以任何比例混合的混合物。
5.根据权利要求1所述的光解方法,其特征在于,所述的金属酞菁为通式(I)的化合物,所述的金属卟啉为通式(II)的化合物;
其中,M独立的选自Fe、Ru、Co、Ni和V;
R1、R2和R3各自独立的选自Cl、F、NO2、SO3H、H、CnH2n+1,其中:n为1~5的整数;
Y1、Y2、Y3、Y4各自独立选自Cm,其中m为0或2。
6.根据权利要求1所述的光解方法,其特征在于,所述的金属酞菁或金属卟啉的用量为反应体系总重量的0.01%~10%。
7.根据权利要求1所述的光解方法,其特征在于,所述的氧化剂为分子氧、空气氧或过氧化物。
8.根据权利要求7所述的光解方法,其特征在于,所述的过氧化物为通式(Ⅲ)或通式(Ⅳ)的化合物;
其中:
R4和R5各自独立的选自CpH2p+1、CpH2p-1或通式(Ⅴ)的化合物;
R6、R7、R8、R9和R10各自独立的选自CqH2q+1、F、Cl、Br、SO3H、NO2、H或OCH3;
p为1~6的整数;
q为0~5的整数。
9.根据权利要求8所述的光解方法,其特征在于,所述的过氧化物与苯并噻吩类化合物的摩尔比为1:1~20:1。
10.根据权利要求1所述的光解方法,其特征在于,所述的苯并噻吩类化合物为通式(Ⅵ)或通式(Ⅶ)的化合物;
其中R11、R12、R13、R14、R15和R16各自独立的选自H、OCH3、COOCH3或CrH2r+1,其中r为1~5的整数。
说明书
技术领域
本发明涉及一种用于氧化脱硫的光解反应方法,尤其涉及一种苯并噻吩化合物的氧化光解反应方法。
背景技术
含硫石油产品经燃烧会释放SOx化合物,对环境造成危害。随着环保相关法规的日益严格与规范,石油产品的深度脱硫正在受到广泛关注。传统的工业脱硫方法是加氢脱硫(HDS),然而苯并噻吩,特别是二苯并噻吩对加氢脱硫有很大抗拒性,达到深度脱硫效果,需要更为苛刻的条件和特殊催化剂,兼具温和和经济性的催化加氢脱硫方法正在紧急地研究中。范煜等人1发明反相微乳核加氢催化剂制备技术,该催化剂在反应压力1.5MPa、反应温度230℃、氢油体积比为300的条件下表现出优异的加氢催化效果。磷钼类催化剂是较早使用的一种HDS催化剂,新研究表明调整制备条件能进一步提高该类催化剂的催化效果2。2013年JACS发表一种纳米纤维束负载的钼类催化剂,在290℃,总压力5MPa下,能够催化加氢达到燃油超清洁深度脱硫效果3。S.H.Moon等人在温度470℃,氢气压力为4MPa时,经过一定时间,二苯并噻吩(DBT)的转化率为60%4。更为先进的催化剂,例如:Zdenek Vít等人用含贵金属铂与钯的物质为催化剂,在一定条件下对噻吩进行加氢脱硫反应,能达到一定的脱硫效果5;T.A.Zepeda等人用含稀有金属镓的物质为催化剂,在一定条件下,对二苯并噻吩进行加氢脱硫反应能达到一定的脱硫效果6;Jose′A.Rodriguez等人用含金物质做催化剂,对噻吩进行加氢脱硫反应的研究7;Gerard Parkin等人8以Mo(PMe3)6为催化剂,对苯并噻吩进行加氢脱硫反应,可以达到一定的脱硫效果;Y.Kanda等人用负载铂为催化剂,在一定条件下对噻吩进行加氢脱硫,转化率为接近80%9。虽然这些方法都进行了科学上的有益探索,但是这些苛刻的反应条件、巨大的氢源需求以及硫化物对催化剂的毒性问题都使HDS难以真正达到烃类燃料的无硫化。
与加氢脱硫采用相反反应方式的氧化脱硫备受青睐。氧化脱硫采用氧化的方式,将苯并噻吩及其衍生物类化合物氧化成相应的亚砜或砜,该类化合物极性增加,易于采用物理方式进行分离,达到脱硫目的,例如:Mingyuan Zhu等人10主要含硅的比较温和的催化剂在一定条件下,对DBT进行氧化来达到脱硫的目的,脱硫转化率能达到接近100%;Juan Zhang等人11用酞菁钴做催化剂,在温和条件下,利用空气氧化DBT成二苯并噻吩砜(DBTO2)来达到脱硫效果,结果显示脱硫率能达到80%以上;Lino A.Gonzalez等人12利用廉价、环保的活性炭和超声波技术,在一定条件下,进行氧化脱硫,能达到好的效果;Fa-tang Li等人13利用来源丰富的Fe2O3做催化剂,在光照条件下,DBT的脱硫转化率能达到92.3%。虽然这些方式都能达到深度脱硫效果,且比加氢脱硫的成本更低,对反应装置的要求也更低,但是,氧化脱硫并不是剔除方式脱硫,即亚砜和砜被分离出来时,携带走含CH的部分,造成烃类物质的损失。
氧化断裂苯并噻吩及其衍生物C-S键非常困难,报道较少,仅有的几篇报道几乎都是采用强碱或高温裂解方式。Chunshan Song等人14以30MgO/SiO2为催化剂,二苯并噻吩砜和苯并噻吩砜为底物,在450℃高温下分解二苯并噻吩砜,分解率能达到77%;苯并噻吩砜更为活泼,在400℃时分解率能达到接近97%。这种方法能达到比较高的C-S键断裂产物的产率,但是由于反应温度过高,对设备的要求很高,存在能耗太大的问题;Thomas G.Squires等人15以DBTO2为底物,均三甲基苯为溶剂,加入强碱和冠醚做为催化剂在高温的条件下得到了30%的碳硫键断裂的产物(亚磺酸内酯)。此方法在一定程度上达到了断裂C-S键的目的,但是产率却比较低,还有大部分的原料残留。中国专利CN101301627B也采用金属酞菁和金属卟啉催化剂16,在分子氧作用下,对苯并噻吩及其衍生物进行氧化,所得氧化产物是苯并噻吩类的砜,如上所述此类砜极为稳定,难以达到C-S键断裂。
发明内容
鉴于目前氧化断裂苯并噻吩及其衍生物反应中C-S键难以断裂的问题,本发明的目的在于提供一种在温和的条件下进行苯并噻吩类化合物C-S键氧化断裂的新方法。本发明以金属酞菁或金属卟啉作为催化剂,氧化苯并噻吩类形成亚砜类中间产物,其经常温和常压下光照反应,达到苯并噻吩类化合物C-S键的选择性断裂,该方法反应条件温和,能够为经济性和深度性脱硫提供基础反应方法。
本发明是通过下面所述的技术方案来实现的:
一种用于氧化脱硫的苯并噻吩类化合物的光解方法,该方法包括苯并噻吩类化合物在氧化剂以及金属酞菁或金属卟啉存在下,经过光照反应达到C-S键断裂的反应。
在上述所述的技术方案中,所述的光照反应优选在10~50℃、常压下进行。所述光照反应温度更优选30~40℃,最优选35℃。所述光照反应时间可以根据苯并噻吩类化合物C-S键断裂的分解产物的转化率来调整,在上述反应条件下,反应1~12小时可达到较好的C-S键断裂转化率。
在上述所述的所有技术方案中,所述光照反应中采用的光是指波长在200~600nm范围内的光,更优选为300~500nm。所述的光可以为可见光或紫外光。
在上述所述的所有技术方案中,所述的光照反应的反应溶剂为C1-3醇化合物、C6-20烃化合物或二者以任何比例混合的混合物,其中所述的C6-20烃化合物优选为C6-20烷烃、C6-20环烷烃,更优选C6-12烷烃、C6-12环烷烃。
在上述所述的金属酞菁或金属卟啉可以采用本领域常用的可以实现本发明的现有金属酞菁或金属卟啉。本发明可以优选采用通式(I)所示的金属酞菁或通式(II)所示的金属卟啉;
其中,式(I)和(II)中所述的M为过度金属,M选自Fe、Ru、Co、Ni和V,M优选为Fe、Ru、Co,更优选为Fe;
R1、R2和R3各自独立的选自Cl、F、NO2、SO3H、H或CnH2n+1,其中n为1~5的整数;优选的R1、R2和R3各自独立的选自Cl、F、NO2或CnH2n+1,其中n为1~3的整数。
Y1、Y2、Y3、Y4各自独立的选自Cm,其中m为0或2。
本发明中,上述式(I)所述的金属酞菁或式(II)所述的金属卟啉可以购买或采用本领域常规制备方法制备得到,其中m=2时的式(II)所示的金属卟啉可以由Peter K17等公开的方法制备得到。
在上述所述所有技术方案中,所述的金属酞菁或金属卟啉的用量为反应体系总重量的0.01%~10%,更优选为0.1%~1%。
在上述所述所有技术方案中,所述的氧化剂为分子氧、空气氧或过氧化物,其中过氧化物优选采用通式(Ⅲ)或通式(Ⅳ)的化合物:
其中:
R4选自CpH2p+1、CpH2p-1或通式(Ⅴ)的化合物,其中p为1~6的整数,优选p为4~6的整数;
R5选自CpH2p+1、CpH2p-1或通式(Ⅴ)的化合物,其中p为1~6的整数,优选p为1或2;
R6、R7、R8、R9和R10各自独立的选自CpH2p+1、F、Cl、Br、SO3H、NO2、H或OCH3,p为1~6的整数;优选R6、R7、R8、R9和R10各自独立的选自CpH2p+1、F、Cl、Br、NO2,p为1~6的整数;更优选R6、R7、R8、R9和R10各自独立的选自Cl或NO2;
q为0~5的整数,优选q为0或1。
在上述所述所有技术方案中,所述的过氧化物与苯并噻吩类化合物的摩尔比为1:1~20:1,优选为4:1~8:1。在上述技术方案中,氧化剂为分子氧、空气氧时,分子氧或空气在常压下按照一定的速度通入反应液中,通入速度不予具体限定,一般为气泡从反应液底部连续冒泡程度即可。本领域技术人员根据需要可以通入带有一定压力的分子氧或空气,或密闭反应容器,使反应在一定的气压下进行,可以达到缩短反应时间的技术效果。
在上述所述所有技术方案中,所述的苯并噻吩类化合物优选为通式(Ⅵ)或通式(Ⅶ)的化合物;
其中,R11、R12、R13、R14、R15和R16各自独立的选自H、OCH3、COOCH3或CrH2r+1,其中r为1~5的整数,r优选1或2。
本发明所述的苯并噻吩类化合物的光解反应,反应机理为:(1)苯并噻吩类化合物先在氧化剂、金属酞菁或金属卟啉作用下形成亚砜,所述亚砜为整体反应过程的重要中间产物,与金属酞菁或金属卟啉形成配合物,能够吸收特定的紫外光或可见光;(2)与金属酞菁或金属卟啉形成配合物的亚砜在特定波长的光和金属酞菁或金属卟啉催化作用下,进行光解反应,形成C-S键断裂产物亚磺酸内酯,亚磺酸内酯不稳定,继续光照氧化成2-羟基联苯和其它磺酸和硫酸等极性大的物质。以卟啉铁为例本发明所述苯并噻吩类化合物的光解反应的反应原理如下:
基于上述本发明光解反应方法的反应原理,在具体实施时,本发明所述的苯并噻吩类化合物C-S键断裂的整体光解反应,可以采用分步反应法或连续反应法来完成。所述分步反应法是指:(1)将苯并噻吩类化合物、金属酞菁或金属卟啉以及氧化剂,在常温常压下混合于溶剂中,制得中间产物苯并噻吩的亚砜类化合物;(2)从步骤(1)的反应液中分离得到中间产物,中间产物中加入与步骤(1)相同的溶剂和金属酞菁或金属卟啉,然后进行光照反应,得到苯并噻吩类化合物C-S键断裂的分解产物。
所述的连续反应法是指:将苯并噻吩类化合物、金属酞菁或金属卟啉、氧化剂,在常温常压下混合于溶剂中,混合液直接进行光照反应,得到苯并噻吩类化合物C-S键断裂的分解产物。
本发明的有益效果:
①本发明的方法在常温常压的温和条件下,氧化石油产品中最为稳定的一类硫化物-苯并噻吩类化合物,达到苯并噻吩类化合物分子中C-S键氧化断裂,得到极性更大的含硫化合物和不含硫的烃类氧化物,在邻氯四苯基卟啉铁为催化剂时C-S键断裂转化率达到90%以上,克服了目前苯并噻吩类化合物分子中C-S键氧化断裂反应需要高温、高压和特殊催化剂,对设备的要求高,产率低等问题。
②本发明的方法中,苯并噻吩类化合物C-S键氧化断裂生成的主产物为不含硫的烃类氧化物,解决了目前氧化脱硫并不是剔除方式脱硫,即以亚砜和砜形式分离出来,携带走含CH的部分,造成烃类物质的损失的问题;本发明中苯并噻吩类化合物C-S键氧化断裂生成的其它氧化产物为含硫的磺酸或硫酸类化合物,由于极性较之前报道的氧化产物砜和亚砜的极性大很多,可自行沉淀过滤分离,极大减少过去因吸附等方式分离亚砜或砜所造成的烃类损失。
附图说明
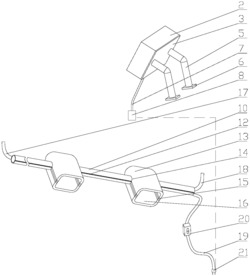
图1为本发明所述的用于氧化脱硫的苯并噻吩类化合物光解反应装置图。
附图标号:1、三口反应器,2、灯,3、冷凝管,4、反应区,5、灯开关
具体实施方式
下述非限制性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。下述实施例中,如无特殊说明,所使用的实验方法均为常规方法,所用材料、试剂等均可从生物或化学公司购买。
实施例1
称取30mg(0.15mmol)二苯并噻吩(DBT),5mg邻氯四苯基卟啉铁(即通式(II)中M=Fe,R1=Cl,R2=R3=H,Y1=Y2=Y3=Y4=Cm,m=0),溶于30mL甲醇溶液中,慢慢滴加54mg(0.9mmol)的过氧化叔丁醇(TBHP)于反应混合液中,混合液在250w高压汞灯的照射下搅拌反应2.5小时,用冷却水循环保持反应温度在36℃。经色谱柱分离和核磁共振检测,反应产物中二苯并噻吩C-S键断裂的转化率为95.21%。
上述反应优选采用如图1所示的反应装置中进行,具体为:三口反应器1,由中心口装入U型冷凝管3,冷凝管的U型区内设置灯2,由灯开关5来控制灯2的开和关。三口反应器1中,U型冷凝管3和灯2以外的部分为反应区4。反应时:将反应原料(如实施例1中,二苯并噻吩,邻氯四苯基卟啉铁和甲醇)放入反应区4中,慢慢滴加氧化剂(过氧化叔丁醇)于反应混合液中,混合液在灯(高压汞灯)2的照射下搅拌反应2小时,通过冷凝管3循环冷凝水,使反应液温度保持在设定的温度。
下述实施例2~8也采用图1的装置完成的,因具体反应时的装置操作方式同实施例1,在下述实施例2~8中不再重复陈述。
实施例2
称取30mg4,6-二甲基二苯并噻吩(DMDBT),5mg邻氯四苯基卟啉铁,溶于30mL甲醇溶液中,慢慢滴加1.81g H2O2(30%水溶液)于反应混合液中,混合液在250w高压汞灯的照射下搅拌反应2.5小时,用冷却水保持反应温度在36℃。经高效液相色谱检测,4,6-二甲基二苯并噻吩100%转化,经色谱柱分离和核磁共振检测,4,6-二甲基二苯并噻吩C-S键断裂的转化率为93.27%。
实施例3
称取30mg二苯并噻吩(DBT),5mg酞菁铁(即通式(I)中M=Fe,R1=R2=R3=H,),溶于30mL甲醇溶液中,慢慢滴加0.72mg TBHP于反应混合液中,混合液在250w高压汞灯的照射下搅拌反应2.5小时,用冷却水循环保持反应温度在25℃。经色谱柱分离和核磁共振检测,反应产物中二苯并噻吩C-S键断裂的转化率为67.20%。
实施例4
称取30mg二苯并噻吩(DBT),5mg邻氯四苯基卟啉铁,溶于30mL十氢化萘和10mL甲醇混合溶液中,慢慢滴加0.72mg TBHP于反应混合液中,在250w高压汞灯的照射下搅拌反应2.5小时,用冷却水循环保持反应温度在36℃。经色谱柱分离和核磁共振检测,反应产物中二苯并噻吩C-S键断裂的转化率为97.01%。
实施例5
称取30mg二苯并噻吩(DBT),5mg邻氯四苯基卟啉铁,溶于乙醇溶液中,通入氧气于反应混合液底部,保持氧气泡连续上升,在250w高压汞灯的照射下搅拌反应2.5小时,用冷却水循环保持反应温度在36℃。经色谱柱分离和核磁共振检测,反应产物中二苯并噻吩C-S键断裂的转化率为92.36%。
实施例6
称取30mg苯并噻吩(BT),5mg邻氯四苯基卟啉铁,溶于乙醇溶液中,通入氧气于反应混合液底部,保持氧气泡连续上升,在250w高压汞灯的照射下搅拌反应2.5小时,用冷却水循环保持反应温度在36℃。经色谱柱分离和核磁共振检测,反应产物中二苯并噻吩C-S键断裂的转化率为90.36%。
实施例7
称取30mg(0.15mmol)二苯并噻吩(DBT),5mg邻氯四苯基卟啉钴(即通式(II)中M=Co,R1=Cl,R2=R3=H,Y1=Y2=Y3=Y4=Cm,m=0),溶于30mL甲醇溶液中,慢慢滴加54mg(0.9mmol)的过氧化叔丁醇(TBHP)于反应混合液中,混合液在250w高压汞灯的照射下搅拌反应2.5小时,用冷却水循环保持反应温度在36℃。经色谱柱分离和核磁共振检测,反应产物中二苯并噻吩C-S键断裂的转化率为15.00%。
实施例8
称取30mg(0.15mmol)二苯并噻吩(DBT),5mg邻氯二苯炔基,邻氯二苯基卟啉铁(即通式(II)中M=Fe,R1=Cl,R2=R3=H,Y1=Y3=Cm,m=0;Y2=Y4=Cm,m=2),溶于30mL甲醇溶液中,慢慢滴加54mg(0.9mmol)的过氧化叔丁醇(TBHP)于反应混合液中,混合液在250w高压汞灯的照射下搅拌反应2.5小时,用冷却水循环保持反应温度在36℃。经色谱柱分离和核磁共振检测,反应产物中二苯并噻吩C-S键断裂的转化率为42.50%。
参考文献:
[1]范煜,夏少青,李国旗等.一种汽油深度加氢脱硫催化材料及催化剂的制备方法.中国专利CN102941125B.
[2]R Wang,K.J.Smith.Preparation of Unsupported NiMoP Catalysts for4,6-Dimethyldibenzothiophene Hydrodesulfurization.Catalysis Letters.2014.
[3]T Tang,et al.Design and Synthesis of Metal Sulfide Catalysts Supported on Zeolite Nanofiber Bundles with Unprecedented Hydrodesulfurization Activities.J.Am.Chem.Soc.2013,135,11437-11440.
[4]J.H.Koh,J.J.Lee,H.Kim,A.Cho,S.H.Moon.Correlation of the deactivation of CoMo/Al2O3 in hydrodesulfurization with surface carbon species.Applied Catalysis B:Environmental86(2009)176-181.
[5]Z.Vít,D.Gulková,L. M.Boaro.Effect of Catalyst Precursor and Its Pretreatment on the Amount of Β-Pd Hydride Phase and Hds Activity of Pd-Pt/Silica-Alumina[J].Applied Catalysis B:Environmental,2014,146(0):213-220.
[6]T.A.Zepeda,B.Pawelec,J.N.Díaz de León,J.A.de los Reyes,A.Olivas.Effect of gallium loading on the hydrodesulfurization activity of unsupported Ga2S3/WS2 catalysts.Applied Catalysis B:Environmental111-112(2012)10-19.
[7]Jose′A.Rodriguez,Francesc Illas etc.Desulfurization of Thiophene on Au/TiC(001):Au-C Interactions and Charge Polarization.J.Am.Chem.Soc.2009,131,8595-8602.
[8]D.Buccella,K.E.Janak,G.Parkin.Reactivity of Mo(PMe3)6towards Benzothiophene and Selenophenes:New Pathways Relevant to Hydrodesulfurization.J.Am.Chem.Soc.2008,130,16187-16189.
[9]Y.Kanda,T.Aizawa,T.Kobayashi,Y.Uemichi,S.Namba,M.Sugioka.Preparation of highly active AlSBA-15-supported platinum catalyst for thiophene hydrodesulfurization.Applied Catalysis B:Environmental77(2007)117-124.
[10]M Zhu,G Luo,L Kang and B Dai.Novel catalyst by immobilizing a phosphotungstic acid on polymer brushes and its application in oxidative desulfurization.RSC Adv.,2014,4,16769-16776.
[11]J Zhang,J Li,T Ren,Y Hu,J Ge and D Zhao.Oxidative desulfurization of dibenzothiophenebased on air and cobalt phthalocyanine in an ionic liquid.RSC Adv.2014,4,3206-3210.
[12]L.A.Gonzalez,P.Kracke,W.H.Green,J.W.Tester,L.M.Shafer,and M.T.Timko.Oxidative Desulfurization of Middle-Distillate Fuels Using Activated Carbon and Power Ultrasound.Energy Fuels2012,26,5164-5176.
[13]F Li,Y Liu,Z Sun,Y Zhao,R Liu,L Chen and D Zhao.Photocatalytic oxidative desulfurization of dibenzothiophene under simulated sunlight irradiation with mixed-phase Fe2O3 prepared by solution combustion.Catal.Sci.Technol.,2012,2,1455-1462.
[14]R.Sundararaman,C Song.Catalytic Decomposition of benzothiophenic and dibenzothiophenic sulfones over MgO-based catalysts.Applied Catalysis,B:Environmental(2014),148-149,80-90.
[15]T.G.Squires,C.G.Venier,B.A.Hodgson et al.Preparation,characterization,and flash vacuum pyrolysis of dibenz[c,e][1,2]oxat hiin6-oxide(biphenylene sultine)J.Org.Chem.,1981,46(11):2376-2379.
[16]周新锐,张珊珊;李娟.仿生催化氧气氧化噻吩类化合物的方法.中国专利CN101301627B.
[17]P.K.Goldberg,T.J.Pundsack,K.E.Splan.J.Phys.Chem.A2011,115,10452-10460.
一种用于氧化脱硫的苯并噻吩类化合物光解反应方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0