

IPC分类号 : C01B39/00,B01J29/03,C07C409/22,C07C409/20






专利摘要
本发明涉及催化材料合成领域,具体提供了一种介孔硅材料及其合成方法,该方法包括:(1)将有机硅源、酸源与表面活性剂进行水解得到混合物A,将得到的混合物A进行第一晶化;(2)将第一晶化后的物料与无机硅源混合得到混合物B,将得到的混合物B进行第二晶化。本发明提供了按照本发明的方法合成得到的介孔硅材料。本发明提供了本发明的介孔硅材料在氧化反应中的应用。本发明提供了一种氧化环酮的方法。本发明提供的合成介孔硅材料的方法,能够利用相对廉价的无机硅源例如硅胶或硅溶胶为部分甚至主要硅源,减少了有机硅酯的用量,大幅度提高了合成效益。
权利要求
1.一种合成介孔硅材料的方法,该方法包括:
(1)将有机硅源、酸源与表面活性剂进行水解得到混合物A,将得到的混合物A进行第一晶化;
(2)将第一晶化后的物料与无机硅源混合得到混合物B,将得到的混合物B进行第二晶化。
2.根据权利要求1所述的方法,其中,第二晶化的温度比第一晶化的温度高10-50℃。
3.根据权利要求1所述的方法,其中,第二晶化的时间比第一晶化的时间长5-24h。
4.根据权利要求1-3中任意一项所述的方法,其中,第一晶化的温度为50-150℃,第一晶化的时间为1-12h;第二晶化的温度为60-200℃,第二晶化的时间为6-36h。
5.根据权利要求1-3中任意一项所述的方法,其中,以SiO2计,有机硅源与无机硅源的用量摩尔比为0.01-0.95:1。
6.根据权利要求1-3中任意一项所述的方法,其中,混合物A中有机硅源的水解率为10-100%。
7.根据权利要求1-3中任意一项所述的方法,其中,步骤(1)中,有机硅源、酸源、表面活性剂和水的用量摩尔比为1:(0.15-0.35):(0.05-0.25):(25-150),其中,有机硅源以SiO2计、酸源以H+计。
8.根据权利要求1-3中任意一项所述的方法,其中,步骤(1)中,所述有机硅源为选自式I所示的含硅化合物中的一种或多种;
式I中,R1、R2、R3和R4各自为C1-C4的烷基。
9.根据权利要求1-3中任意一项所述的方法,其中,所述表面活性剂为非离子表面活性剂;所述酸源为有机酸和/或无机酸,所述无机硅源为硅溶胶和/或硅胶。
10.根据权利要求1-3中任意一项所述的方法,其中,该方法还包括:将第二晶化所得产物过滤、洗涤得到固体,将所得固体干燥或不干燥后进行焙烧。
11.权利要求1-10中任意一项所述的方法得到的介孔硅材料。
12.权利要求11所述的介孔硅材料在氧化反应中的应用。
13.一种氧化环酮的方法,该方法包括:在含氧气体存在条件下,将环酮与催化剂接触,其特征在于,所述催化剂含有权利要求11所述的介孔硅材料。
14.根据权利要求13所述的方法,其中,所述含氧气体含有臭氧。
说明书
技术领域
本发明涉及一种介孔硅材料的合成方法和由该方法合成得到的介孔硅材料及其在氧化反应中的应用,以及一种氧化环酮的方法。
背景技术
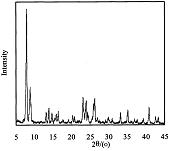
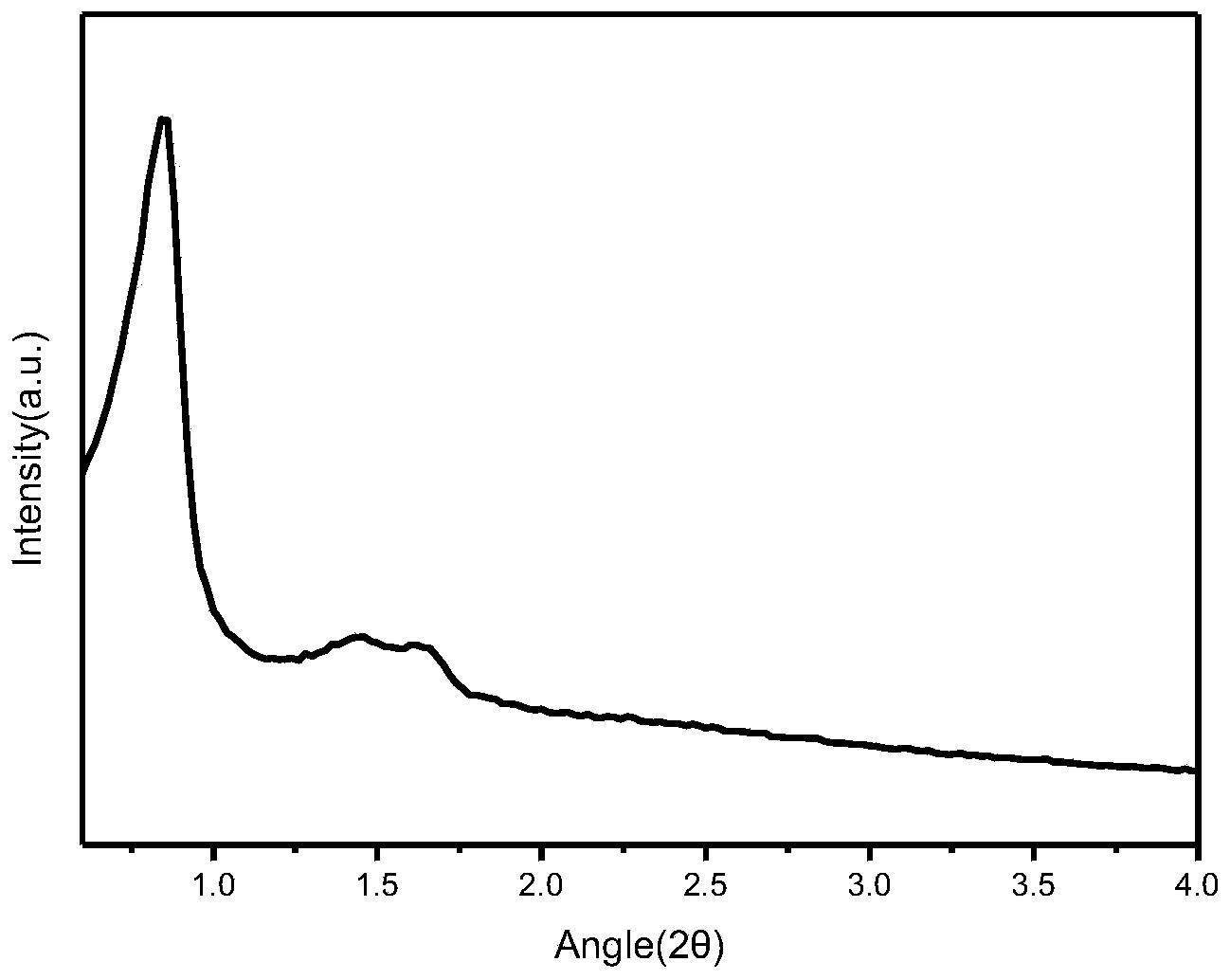
赵东元等首次报道了SBA-15系列的介孔分子筛及其合成方法(Science,1998,279,548),该系列材料具有均匀的颗粒度和规整的介孔结构,吸引了相关学术界的极大关注,为大分子的催化、分离等方面带来了希望。它的介孔结构特征是其X射线衍射谱图在2θ为0.8°附近、1.5°附近、1.7°附近处有衍射峰。但现有方法合成出的硅分子筛SBA-15其合成效率和产品收率等方面还有待进一步的改进。
发明内容
本发明的目的是针对现有合成介孔硅材料方面的不足,提供一种合成介孔硅材料的方法。
本发明的发明人经过大量的研究发现,在介孔硅材料的制备过程中,若将有机硅源、表面活性剂、酸源进行混合并水解,将水解得到的混合物转入密封反应釜中晶化,晶化后往晶化体系中加入无机硅源并混匀,再将混合物继续在密封反应釜中晶化,这样不仅节省制备成本,而且这样合成得到的介孔硅材料,令人意外的是,分子筛收率和相对结晶度均较高,更令人意外的是,其在5-10nm之间的孔径分布占总孔径分布的百分数更高,且分子筛催化氧化活性高。基于此,完成了本发明。
为实现前述目的,本发明的第一方面,本发明提供了一种合成介孔硅材料的方法,该方法包括:
(1)将有机硅源、酸源与表面活性剂进行水解得到混合物A,将得到的混合物A进行第一晶化;
(2)将第一晶化后的物料与无机硅源混合得到混合物B,将得到的混合物B进行第二晶化。
本发明的第二方面,本发明提供了按照本发明的方法合成得到的介孔硅材料。
本发明的第三方面,本发明提供了本发明的介孔硅材料在氧化反应中的应用。
本发明的第四方面,本发明提供了一种氧化环酮的方法,该方法包括:在含氧气体存在条件下,将环酮与催化剂接触,所述催化剂含有本发明所述的介孔硅材料。
本发明提供的合成介孔硅材料的方法,能够利用相对廉价的无机硅源例如硅胶或硅溶胶为部分甚至主要硅源,减少了有机硅酯的用量,大幅度提高了合成效益,并且按照本发明的方法合成得到的介孔硅材料相对结晶度高,且在5-10nm之间的孔径分布占总孔径分布的百分数更高。而且在探针反应例如在臭氧氧化环酮反应中,其表现出催化活性高以及过氧化环酮选择性高的特点。
本发明的其他特征和优点将在随后的具体实施方式部分予以详细说明。
具体实施方式
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
如前所述,本发明提供了一种合成介孔硅材料的方法,该方法包括:
(1)将有机硅源、酸源与表面活性剂进行水解得到混合物A,将得到的混合物A进行第一晶化;
(2)将第一晶化后的物料与无机硅源混合得到混合物B,将得到的混合物B进行第二晶化。
根据本发明的方法,优选步骤(1)中,将有机硅源和表面活性剂各自或同时加入到酸源水溶液中进行水解。
根据本发明的方法,只要按照前述技术方案合成介孔硅材料即可实现本发明的目的,针对本发明,为了进一步提高介孔硅材料的物化性能,优选第二晶化的温度比第一晶化的温度高10-50℃,更优选第二晶化的温度比第一晶化的温度高20-40℃,如此可以取得更好效果,例如从下述的表1数据可以看出,晶化温度的差别为40℃的情况下(实施例1),样品的晶粒尺寸、相对结晶度以及催化性能均优于晶化温度没有差别的情况(实施例6)。
根据本发明的方法,只要按照前述技术方案合成介孔硅材料即可实现本发明的目的,针对本发明,为了进一步提高介孔硅材料的物化性能,优选第二晶化的时间比第一晶化的时间长5-24h,更优选第二晶化的时间比第一晶化的时间长6-12h,这样可以取得更好的效果,例如从下述表1数据可以看出,晶化的时间差别为6h的情况下(实施例1),样品的晶粒尺寸、相对结晶度以及催化性能均优于晶化时间没有差别的情况(实施例8)。
根据本发明的方法,只要按照前述技术方案合成介孔硅材料即可实现本发明的目的,针对本发明,为了进一步提高介孔硅材料的物化性能,优选第一晶化的温度为50-150℃,优选为80-120℃。
根据本发明的方法,只要按照前述技术方案合成介孔硅材料即可实现本发明的目的,针对本发明,为了进一步提高介孔硅材料的物化性能,优选第一晶化的时间为1-12h,优选为5-12h。
根据本发明的方法,只要按照前述技术方案合成介孔硅材料即可实现本发明的目的,针对本发明,为了进一步提高介孔硅材料的物化性能,优选第二晶化的温度为60-200℃,优选为100-180℃。
根据本发明的方法,只要按照前述技术方案合成介孔硅材料即可实现本发明的目的,针对本发明,为了进一步提高介孔硅材料的物化性能,优选第二晶化的时间为6-36h,优选为15-20h。
根据本发明的方法,只要按照前述技术方案合成介孔硅材料即可实现本发明的目的,针对本发明,为了进一步提高介孔硅材料的物化性能,优选以SiO2计,有机硅源与无机硅源的用量摩尔比为0.01-0.95:1,更优选有机硅源与无机硅源的用量摩尔比0.05-0.5:1,进一步优选有机硅源与无机硅源的用量摩尔比0.1-0.5:1。
根据本发明的方法,优选混合物A中有机硅源的水解率为10-100%,进一步优选为50-90%,更优选为60-80%。如此可以进一步提高合成得到的介孔硅材料的物化性能。
根据本发明的方法,只要按照前述技术方案合成介孔硅材料即可实现本发明的目的,针对本发明,为了进一步提高介孔硅材料的物化性能,优选步骤(1)中,有机硅源、酸源、表面活性剂和水的用量摩尔比为1:(0.05-0.5):(0.01-0.2):(5-200),其中,有机硅源以SiO2计、酸源以H+计,更优选有机硅源:酸源:表面活性剂:水=1:(0.15-0.35):(0.05-0.25):(25-150)。
根据本发明的方法,所述有机硅源可以为各种在水解缩合反应条件下能够形成二氧化硅的含硅化合物。具体地,所述有机硅源可以为选自式I所示的含硅化合物中的一种或多种,
式I中,R1、R2、R3和R4各自为C1~C4的烷基,包括C1~C4的直链烷基和C3~C4的支链烷基,例如:R1、R2、R3和R4各自可以为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基。
具体地,所述有机硅源可以为正硅酸四甲酯、正硅酸四乙酯、正硅酸四正丙酯和正硅酸四正丁酯中的一种或多种。在本发明的具体实施例中使用的为正硅酸四乙酯或正硅酸甲酯作为实例,但并不因此限制本发明的范围。
根据本发明的方法,所述酸源的种类的可选范围较宽,其可以为有机酸和/或无机酸,其中,无机酸可以为HCl、硫酸、硝酸和磷酸中的一种或多种,所述有机酸可以为C1-C10的有机羧酸,优选为甲酸、乙酸、丙酸和环烷酸中的一种或多种;优选所述酸源为HCl、硫酸、磷酸和乙酸中的一种或多种。在本发明的实施例中使用的酸为硫酸、盐酸作为实例,但并不因此限制本发明的范围。
根据本发明的方法,优选所述表面活性剂为非离子型表面活性剂,其种类的可选范围较宽,针对本发明,优选所述表面活性剂为脂肪醇聚氧乙烯醚型非离子表面活性剂、聚氧乙烯-聚氧丙烯嵌段共聚物型非离子表面活性剂、酯型非离子表面活性剂、烷基醇酰胺非离子表面活性剂、氟碳非离子表面活性剂以及聚乙烯吡咯烷酮非离子表面活性剂中的一种或多种。所述表面活性剂的具体实例可以包括但不限于:脂肪醇聚氧乙烯醚、脂肪醇聚氧丙烯醚、聚氧乙烯-聚氧丙烯嵌段共聚物型表面活性剂、烷基醇酰胺、多元醇酯型表面活性剂、吐温系列表面活性剂、司盘系列表面活性剂、氟碳表面活性剂和聚乙烯吡咯烷酮。具体可以依据需要合成的介孔硅材料的种类进行选择,在本发明的具体实施例中使用的表面活性剂EO20PO70EO20(Pluronic P123),简称为P123作为实例,但并不因此限制本发明的范围。
根据本发明的方法,所述无机硅源的种类的可选范围较宽,针对本发明,优选所述无机硅源为硅溶胶和/或硅胶,本发明中所述硅胶或硅溶胶可以是各种形式各种生产方法得到的硅胶或硅溶胶,优选所述无机硅源为硅胶。各种硅胶因其制造方法不同而形成不同的微孔结构,如硅胶根据其孔径的大小分为:大孔硅胶、粗孔硅胶、B型硅胶、细孔硅胶等。本发明对所述的硅胶颗粒的孔径、比表面积和孔容(孔体积)等没有特殊要求,所述硅胶可以商购或按照现有方法制备。在本发明方法中,为了获得更好的技术效果,一般要求或优选硅胶为SiO2的质量百分含量即质量分数大于90%,优选大于95%,更优选大于99%的硅胶;硅溶胶更优选为SiO2的质量分数大于10%,优选大于15%,更优选大于20%的硅溶胶,进一步优选为20-40重量%的硅溶胶。
根据本发明的方法,其中步骤(2)将第一晶化后的物料与无机硅源混合时,所述第一晶化后的物料指第一晶化后冷却降温(无特殊要求,降至室温能打开晶化釜即可)后不经过其它任何处理而得到的物料。
根据本发明的方法,优选该方法还包括:将第二晶化所得产物过滤、洗涤得到固体,将所得固体干燥或不干燥后进行焙烧。
本发明中,所述干燥的条件的可选范围较宽,具体可以参照现有技术进行。针对本发明,优选所述干燥的条件包括:温度为室温至200℃,更优选为80-120℃;时间为1-24h,优选为2-10h。
本发明中,所述焙烧的条件的可选范围较宽,针对本发明优选所述焙烧的条件包括:焙烧的温度为300-800℃,优选为450-550℃;焙烧的时间为2-12h,优选为2-4h;更优选所述焙烧的条件包括:首先在350-600℃于氮气气氛中焙烧0.5-6h,然后在350-600℃于空气气氛中焙烧0.5-12h。
本发明提供了一种按照本发明的方法得到的介孔硅材料。
根据本发明的介孔硅材料具有更高的催化活性。具体地,由本发明的方法制备的介孔硅材料在用作氧化反应的催化剂时,显示出更高的催化活性。
本发明提供了本发明的介孔硅材料在氧化反应中的应用。
本发明提供了一种氧化环酮的方法,该方法包括:在含氧气体存在条件下,将环酮与催化剂接触,所述催化剂含有本发明所述的介孔硅材料。
根据本发明的方法,所述催化剂只要含有本发明的介孔硅材料即可,优选所述催化剂中介孔硅材料的含量为50重量%以上,更优选含量为60-100重量%。在本发明的具体实施例中使用的均为介孔硅材料的含量为100重量%的催化剂,但这并不因此限制本发明的范围。此处的含量指的是不含载体时的催化剂组成。
当所述催化剂为成型体时,所述催化剂还包括载体,其中,载体可以为Al2O3、ZnO、MgO、SiO2、CaO和TiO2、稀土氧化物RE2O3(RE为La、Ce、Y或Nd等)。
本发明中,所述催化剂中,除包括介孔硅材料外,还可以含有其他常用的用于氧化反应的催化剂。
根据本发明的一种优选的实施方式,优选所述催化剂为介孔硅材料,更优选氧化环酮的反应条件包括:温度为0-180℃、压力为0.1-3.0MPa、环酮、含氧气体与任选的溶剂的摩尔比为1:0.1-10:1-150。
根据本发明的一种优选的实施方式,优选所述含氧气体含有臭氧,更优选所述含氧气体含有臭氧和氧气,在具体使用过程中,可以使用稀释气体将所述含氧气体进行稀释,所述稀释气体可以为氮气、氩气、氦气、氖气等惰性气体,也可以为二氧化碳、空气等。
根据本发明的一种优选的实施方式,优选环酮、含氧气体与任选的溶剂的摩尔比为1:0.2-5:1-100,反应温度优选为20-160℃,反应压力优选为0.3-2.5MPa。
本发明提供的氧化环酮的方法中,所说的溶剂优选选自水或甲醇、乙醇、正丙醇、异丙醇、叔丁醇、异丁醇等醇类或丙酮、丁酮等酮类或乙腈等腈类或甲酸、乙酸等羧酸类或它们的混合,优选为丙酮、水或它们的混合。
本发明提供的氧化环酮的方法中,所述环酮为环己酮、环戊酮和甲基环己酮中的一种或多种。
下面通过实施例对本发明作进一步说明,但并不因此限制本发明的内容。
本发明中,对比例和实施例中所用原料均为市售,其中硅胶(小球)为青岛硅胶厂产品,SiO2的质量分数大于95%,硅胶A基本质量参数如下:平均孔径2.6nm,比表面积680㎡/g,孔容0.38ml/g;硅胶B基本质量参数如下:平均孔径5.3nm,比表面550m2/g,孔容为0.71ml/g。
如无特别说明,所用试剂为分析纯试剂,均来自国药集团化学试剂有限公司产品。
实施例样品的X-射线衍射(XRD)晶相图测定在Siemens D5005型X-射线衍射仪上进行,以样品与基准样品衍射特征峰的衍射强度(峰高)的比值来表示样品相对于基准样品的结晶度,这里以对比例1样品为基准样品,其结晶度计为100%,各样品的相对结晶度数据见表1,样品的比表面积和孔径及其分布数据在美国Micromeritics公司的ASAP2405静态氮吸附仪上进行测定;各样品的相对结晶度数据、比表面积和孔径分布数据等见表1。其中,孔径分布百分比数据为样品在5-10nm之间的孔径分布占总孔径分布的百分数。
本发明中,各样品的收率指实际得到的产品质量与理论计算质量(以二氧化硅计)的百分比,数据见表1。
以下实施例中,在混合过程中,依据需要加水或不加水,若其中的投料能够满足水的投料要求,则无需加水,若不满足,则需额外加入水。
对比例和实施例中,有机硅源水解量通过气相色谱法测得。所用气相色谱为Agilent 6890N,配备热导检测器TCD和HP-5的毛细管柱(30m*320μm*25μm)。其中,进样口温度为180℃,柱温为150℃,使用氮气作为载气,载气的流速为25mL/min。具体方法为:取一定量的混合物从气相色谱仪进样口进样,流经色谱柱后利用TCD进行检测并通过外标法进行定量。采用以下公式计算有机硅源水解率:
X有机硅源%=[(mo有机硅源-m有机硅源)/mo有机硅源]×100%
式中,X有机硅源表示有机硅源的水解率;mo有机硅源表示加入的有机硅源的质量;m有机硅源表示未水解的有机硅源的质量。
对比例1
参照文献(Science,1998,279,548)方法合成按照如下步骤合成了SBA-15:将8克EO20PO70EO20(Pluronic P123)溶于250毫升盐酸溶液中(HCl,2M),待P123完全溶解后加入18毫升TEOS(其中含80毫摩尔的SiO2)得到混合物,然后该混合物在40℃恒温搅拌20小时后装入反应釜中,在100℃烘箱中放置48小时。将产物过滤、洗涤、干燥得到SBA-15原粉,原粉经过550℃灼烧5小时除去表面活性剂。经检测,其XRD晶相在小角度即2θ在0.8°附近、1.5°附近、1.7°附近处有衍射峰,表明样品具有类似SBA-15的二维六方介孔结构,其相对结晶度、收率和孔结构参数等数据见表1。
实施例1
先将50克正硅酸四乙酯和EO20PO70EO20(Pluronic P123)加入到盐酸的水溶液中搅拌混合,其中正硅酸四乙酯、盐酸、P123和水的摩尔比为1:0.45:0.15:85,其中有机硅酸酯以SiO2计、酸源以H+计,待硅酯水解(有机硅源水解率为100%)后将混合物转入密封反应釜中在110℃水热晶化12h,冷却后打开反应釜往晶化体系中加入硅胶A并混匀,其中以二氧化硅计,加入的硅胶与有机硅源的摩尔比为1:0.2,再将混合物继续在密封反应釜中于150℃的温度和自生压力下晶化18h,将所得晶化产物过滤、洗涤,并于110℃干燥120分钟,然后在550℃下焙烧3h,获得介孔硅材料A。
经检测,其XRD晶相在小角度即2θ在0.8°、1.5°和1.7°附近处有衍射峰,表明具有类似SBA-15的二维六方中孔结构,其相对结晶度见表1。
实施例2
先将50克正硅酸四甲酯和表面活性剂P123加入到盐酸的水溶液中搅拌混合,其中正硅酸四甲酯、盐酸、P123和水的摩尔比为1:0.15:0.05:25,其中有机硅酸酯以SiO2计、酸源以H+计,待硅酯水解(有机硅源水解率为50%)后将混合物转入密封反应釜中在120℃水热晶化5h,冷却后打开反应釜往晶化体系中加入硅胶B并混匀,其中以二氧化硅计,加入的硅胶与有机硅源的摩尔比为1:0.1,再将混合物继续在密封反应釜中于140℃的温度和自生压力下晶化17h,将所得晶化产物过滤、洗涤,并于110℃烘干120分钟,然后在550℃焙烧3h,获得介孔硅材料B,该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例3
先将50克正硅酸四乙酯和P123加入到硫酸水溶液中搅拌混合,其中正硅酸四乙酯、硫酸、表面活性剂和水的摩尔比为1:0.35:0.25:150,其中有机硅酸酯以SiO2计、酸源以H+计,待硅酯水解(有机硅源水解率为60%)后将混合物转入密封反应釜中在100℃水热晶化12h,冷却后打开反应釜往晶化体系中加入硅胶A并混匀,其中以二氧化硅计,加入的硅胶与有机硅源的摩尔比为1:0.5,再将混合物继续在密封反应釜中于130℃的温度和自生压力下晶化20h,将所得晶化产物过滤、用水洗涤,并于110℃烘干120分钟,然后在550℃焙烧3h,获得介孔硅材料C,该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例4
按照实施例3的方法合成介孔硅材料D,不同的是其中以SiO2计,加入的硅胶与有机硅源的摩尔比为1:0.05。该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例5
按照实施例3的方法合成介孔硅材料E,不同的是,其中正硅酸四乙酯、硫酸、表面活性剂和水的摩尔比为1:0.1:0.05:100。该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例6
按照实施例1的方法合成介孔硅材料F,不同的是加入硅胶前的晶化温度(即第一晶化温度)为150℃。该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例7
按照实施例1的方法合成介孔硅材料G,不同的是加入硅胶后的晶化温度(即第二晶化温度)为100℃。该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例8
按照实施例1的方法合成介孔硅材料H,不同的是加入硅胶后的水热晶化时间(即第二晶化时间)为12h。该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例9
按照实施例1的方法合成介孔硅材料I,不同的是加入硅胶后的水热晶化时间(即第二晶化时间)为6h。该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例10
按照实施例1的方法合成介孔硅材料J,不同的是使用的无机硅源为硅溶胶(二氧化硅含量为40重量%),其所带入的二氧化硅含量与实施例1的硅胶带来的二氧化硅含量相同。该样品经X-射线衍射与实施例1样品A的谱学特征类似。
实施例11
按照实施例1的方法制备介孔硅材料,不同的是,有机硅源的水解率为80%。该样品经X-射线衍射与实施例1样品的谱学特征类似。
对比例2
按照实施例1的方法制备介孔硅材料,不同的是,不进行两次晶化,具体如下:
先将50克正硅酸四乙酯、硅胶A、EO20PO70EO20(Pluronic P123)加入到盐酸的水溶液中搅拌混合,其中正硅酸四乙酯、盐酸、P123和水的摩尔比为1:0.45:0.15:85,其中有机硅酸酯以SiO2计、酸源以H+计,其中以二氧化硅计,加入的硅胶与有机硅源的摩尔比为1:0.2,待硅源水解完全后将混合物转入密封反应釜中在150℃水热晶化30h,将所得晶化产物过滤、洗涤,并于110℃干燥120分钟,然后在550℃下焙烧3h,获得介孔硅材料。
对比例3
按照对比例2的方法进行,不同的是,其中使用的硅胶全部用正硅酸四乙酯代替,其带入的二氧化硅含量与硅胶带入的二氧化硅的含量相同。
测试例
本测试例说明本发明的方法和对比例的方法所得样品用于催化臭氧氧化环己酮反应的效果。
所用臭氧由福建新大陆环保科技有限公司生产的NLO-15型氧气源臭氧发生器提供,臭氧浓度可调,最大体积浓度可达80%(其余为氧气)。
在温度为60℃和压力为0.5MPa下,以臭氧(15%体积比,其余为氧气)为氧化剂,将环己酮、臭氧和溶剂丙酮按照1:1:1的摩尔比进行反应。反应2小时的环己酮转化率和过氧化环己酮选择性结果见表1。
其中:
表1
从表1的结果可以看出:本发明的方法分子筛收率高,其在5-10nm之间的孔径分布占总孔径分布的百分数即孔径分布更高,且本发明的方法制备的样品相对结晶度高,且催化活性明显高于对比例的方法制备的样品,例如特别是对比实施例1与对比例2和3的样品的结果可知,按照本发明的方法使用硅胶代替部分有机硅源使用,经过两次晶化后得到的介孔硅材料的活性远远高于全部使用有机硅源仅通过一次晶化得到的介孔硅材料的活性,由此可见,本发明的方法能够在大量节省有机硅源使用的情况下,仍能提高介孔硅材料的相对结晶度和催化活性。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
介孔硅材料及其合成方法和应用以及一种氧化环酮的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0