
NbO晶体及其制备方法")









专利摘要
本发明公开了一种薄片微纳米(K,Na)NbO3晶体的制备方法,包括如下步骤:(1)将一定量的乙二醇作为溶剂,依次加入一定量的KOH、NaOH、Nb2O5以及表面活性剂,用玻璃棒缓慢搅拌至溶质全部溶解;(2)溶解后的混合液放在磁力搅拌器上搅拌一定时间,倒入水热釜中,在一定温度下水热一段时间后,离心、洗涤、稀盐酸浸泡、烘干得到薄片微纳米(K,Na)NbO3晶体粉末。本发明以乙二醇为溶剂,降低了传统水热法所需的矿化剂浓度,并且提高体系的稳定性,通过改变不同的条件,所得的产物都是以薄片微纳米(K,Na)NbO3晶体粉末为主,该晶体直径为0.2‑2μm,属于微米级。
权利要求
1.一种薄片微纳米(K,Na)NbO
步骤一:将28-32mL的乙二醇作为溶剂,依次加入5.5-6g KOH、1 .5-2g NaOH、1 .5-3gNb
步骤二:溶解后的混合液放在磁力搅拌器上搅拌25-35min,倒入水热釜中,在180-200℃下水热6-8h后,8000r/min离心3min,再用蒸馏水及无水乙醇各洗涤两次,再用稀盐酸浸泡1 .5-2h,在60℃的烘箱中烘干得到薄片微纳米(K,Na)NbO
说明书
技术领域
本发明涉及化学材料制备技术领域,尤其是一种薄片微纳米(K,Na)NbO3晶体及其制备方法。
背景技术
近年来,为解决含铅量超过60%的铅基压电陶瓷所引起的环境问题,越来越多的研究人员将目光投放在绿色材料方面。无铅压电陶瓷以其优良的性能成为了绿色材料领域的热点,(K,Na)NbO3(KNN)属于典型的钙钛矿型铁电体,具有高居里温度,优异的压电性能和合适的禁带宽带,是这类陶瓷中最有前途的环保型候选材料之一。除此之外,开发绿色合成方法也同样占据着重要的位置,应用绿色和可持续化学原理,减少使用危险、昂贵和有毒的前驱体,开发简单、经济、高效的合成方案,从而减少废料的产生,提高材料和能源效率,并使用更安全的替代反应介质代替污染和有毒有机溶剂。
传统的水热法制备合成片状KNN时,多数以模板法为主,制备工艺复杂,且产生的中间相较多,过长的反应时间也导致最后KNN的纯度与厚度都难以控制,除此之外,单纯的水热法合成这些化合物时还需要很高的碱性溶液(10mol/L) 作矿化剂,生产成本较高,不利于批量生产,在实际应用中太过于局限,因此传统的水热法还需进一步的改良。
发明内容
针对上述存在的问题,本发明的目的是提供一种薄片微纳米(K,Na)NbO3晶体及其制备方法。
本发明的技术方案是:一种薄片微纳米(K,Na)NbO3晶体的制备方法,包括如下步骤:
步骤一:将一定量的乙二醇作为溶剂,依次加入一定量的KOH、NaOH、 Nb2O5以及表面活性剂,用玻璃棒缓慢搅拌至溶质全部溶解;
步骤二:溶解后的混合液放在磁力搅拌器上搅拌一定时间,倒入水热釜中,在一定温度下水热一段时间后,离心、洗涤、稀盐酸浸泡、烘干得到薄片微纳米(K,Na)NbO3晶体粉末。
进一步的,所述步骤一具体为将28-32mL的乙二醇作为溶剂,依次加入一 5.5-6gKOH、1.5-2g NaOH、1.5-3g Nb2O5以及0.4-0.6wt%的表面活性剂,用玻璃棒缓慢搅拌至溶质全部溶解。
进一步的,所述步骤二具体为溶解后的混合液放在磁力搅拌器上搅拌 25-35min,倒入水热釜中,在180-200℃下水热6-8h后,8000r/min离心3min,再用蒸馏水及无水乙醇各洗涤两次,再用稀盐酸浸泡1.5-2h,在60℃的烘箱中烘干得到薄片微纳米(K,Na)NbO3晶体粉末。
进一步的,所述步骤二中的表面活性剂为PEG、PVA或CTAB。
进一步的,制得的产物以直径为0.2-1μm薄片微纳米(K,Na)NbO3晶体为主,分散性较好,还有少量的团聚纳米粒杂质附着在薄片上,晶型为钙钛矿型。
与现有技术相比,本发明的有益效果是:
(1)本发明基于工艺简单化的原则,以乙二醇作为溶剂,同时又添加了表面活性剂来增加颗粒表面电势,提高体系的稳定性,在低温下成功的合成以薄片状为主的(K,Na)NbO3晶体粉末,具有较好的分散性,优异的化学稳定性和较窄的粒径分布,期望能得到更广泛的应用。
(2)本发明以乙二醇为溶剂,降低了传统水热法所需的矿化剂浓度,并且提高体系的稳定性,通过改变不同的条件,所得的产物都是以薄片状为主的微纳米(K,Na)NbO3晶体粉末,该晶体直径为0.2-2μm,属于微米级。
附图说明
图1是实施例1中薄片状(K,Na)NbO3(Nb2O5=3g)的扫描电子显微镜图。
图2是实施例1中薄片状(K,Na)NbO3(Nb2O5=3g)的XRD图。
图3是实施例1中薄片状(K,Na)NbO3(Nb2O5=1.5g)的扫描电子显微镜图。
图4是实施例1中薄片状(K,Na)NbO3(Nb2O5=1.5g,表面活性剂为PEG) 的扫描电子显微镜图。
图5是实施例1中薄片状(K,Na)NbO3(Nb2O5=1.5g,表面活性剂为PVA) 的扫描电子显微镜图。
图6是实施例1中薄片状(K,Na)NbO3(Nb2O5=1.5g,表面活性剂为PVA) 的XRD图。
图7是实施例1中薄片状(K,Na)NbO3(Nb2O5=1.5g,表面活性剂为CTAB) 的扫描电子显微镜图。
图8是实施例1中薄片状(K,Na)NbO3(Nb2O5=1.5g,经过酸洗)的扫描电子显微镜图。
具体实施方式
下面结合附图和实施例对本发明作进一步的说明。
实施例1
称量30ml的乙二醇作为溶剂,再依次加入5.88g KOH、1.8g NaOH、3g Nb2O5用玻璃棒缓慢搅拌至溶质全部溶解,再将其放在磁力搅拌器上搅拌30min,之后倒入水热釜中,在180℃的条件下水热8h,8000r/min离心3min,再用蒸馏水及无水乙醇各洗涤两次,在60℃的烘箱中烘干得到薄片状微纳米(K,Na)NbO3晶体粉末。
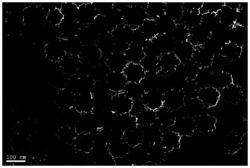
图1为本实施例中(K,Na)NbO3粉末的扫描电子显微镜图,可以看出产物 (K,Na)NbO3是以直径为1-2μm超薄片微纳米(K,Na)NbO3晶体为主,分散性较好;除此之外,四周还散落着纳米颗粒,部分有团聚现象。
图2为本实施例中(K,Na)NbO3粉末的XRD图,可以看出超薄片微纳米 (K,Na)NbO3晶体为钙钛矿型,存在少量杂相,峰窄、峰形尖锐且强度高,表明晶体结构完整。
实施例2
称量30ml的乙二醇作为溶剂,再依次加入5.88g KOH、1.8g NaOH、1.5g Nb2O5用玻璃棒缓慢搅拌至溶质全部溶解,再将其放在磁力搅拌器上搅拌30min,之后倒入水热釜中,在200℃的条件下水热8h,8000r/min离心3min,再用蒸馏水及无水乙醇各洗涤两次,在60℃的烘箱中烘干得到薄片状微纳米(K,Na)NbO3晶体粉末。
图3为本实施例中(K,Na)NbO3粉末的扫描电子显微镜图,可以看出所得产物(K,Na)NbO3是以直径0.8-1.5μm的薄片状为主,除此之外,四周还散落着纳米颗粒,部分有团聚现象,与实施例1相比,在该条件下所得到的片状粒径更小。
实施例3
称量30ml的乙二醇作为溶剂,再依次加入5.88g KOH、1.8g NaOH、1.5g Nb2O5,0.5wt%的表面活性剂PEG,用玻璃棒缓慢搅拌至溶质全部溶解,再将其放在磁力搅拌器上搅拌30min,之后倒入水热釜中,在200℃的条件下水热8h, 8000r/min离心3min,再用蒸馏水及无水乙醇各洗涤两次,在60℃的烘箱中烘干得到超薄片状微纳米(K,Na)NbO3晶体粉末。
图4为本实施例中(K,Na)NbO3粉末的扫描电子显微镜图,可以看出所得产物(K,Na)NbO3与实施例1和实施例2差别较大,但基本还是由片状组成,粒径为1μm左右,略有厚度,除此之外,还有由团聚的纳米颗粒组成的蜂窝状形貌的产物附着在片状产物上。
实施例4
称量30ml的乙二醇作为溶剂,再依次加入5.88g KOH、1.8g NaOH、1.5g Nb2O5、0.4wt%的表面活性剂PVA,用玻璃棒缓慢搅拌至溶质全部溶解,再将其放在磁力搅拌器上搅拌30min,之后倒入水热釜中,在200℃的条件下水热8h, 8000r/min离心3min,再用蒸馏水及无水乙醇各洗涤两次,在60℃的烘箱中烘干得到薄片状微纳米(K,Na)NbO3晶体粉末。
图5为本实施例中(K,Na)NbO3粉末的扫描电子显微镜图,可以看出所得产物(K,Na)NbO3是以直径1.0μm的薄片状为主,除此之外,四周还散落着纳米颗粒,部分有团聚现象。
图6为本实施例中(K,Na)NbO3粉末的XRD图,可以看出薄片微纳米 (K,Na)NbO3晶体为钙钛矿型,存在少量杂相,峰窄、峰形尖锐且强度高,表明晶体结构完整。
实施例5
称量30ml的乙二醇作为溶剂,再依次加入5.88g KOH、1.8g NaOH、1.5g Nb2O5、0.6wt%的表面活性剂CTAB,用玻璃棒缓慢搅拌至溶质全部溶解,再将其放在磁力搅拌器上搅拌30min,之后倒入水热釜中,在200℃的条件下水热8h, 8000r/min离心3min,再用蒸馏水及无水乙醇各洗涤两次,在60℃的烘箱中烘干得到薄片状微纳米(K,Na)NbO3晶体粉末。
图7为本实施例中(K,Na)NbO3粉末的扫描电子显微镜图,可以看出所得产物(K,Na)NbO3是以直径1.0μm的薄片状为主,与实施例4相比,该实施例下的片状产物分散性较好,四周依然还散落着纳米颗粒,部分有团聚现象
实施例6
称量30ml的乙二醇作为溶剂,再依次加入5.88g KOH、1.8g NaOH、1.5g Nb2O5,用玻璃棒缓慢搅拌至溶质全部溶解,再将其放在磁力搅拌器上搅拌30min,之后倒入水热釜中,在200℃的条件下水热8h,8000r/min离心3min,用蒸馏水及无水乙醇各洗涤两次,再用稀盐酸浸泡2h,之后在60℃的烘箱中烘干得到薄片状微纳米(K,Na)NbO3晶体粉末。
图8为本实施例中(K,Na)NbO3粉末的扫描电子显微镜图,制得的产物为直径为0.2-1μm薄片微纳米(K,Na)NbO3晶体,分散性较好,只有少量团聚;与其他实施例相比,经过酸泡的产物里纳米颗粒大量减少,产物晶型依然为钙钛矿型。
对比实施例1-6,其产物都是以薄片微纳米(K,Na)NbO3晶体为主,并且根据 XRD该产物为纯相的KNN,根据检测结果,添加不同量的Nb2O5对产物的粒径略有影响,但基本形貌未发生太大变化;添加不同的表面活性剂对产物的团聚程度有一定的影响,在表面活性剂为CTAB时,其片状产物更加分散,纵横比也相差较大,有少量的纳米颗粒团聚体存在;当表面活性剂为PEG时,其片状产物与其他条件下的产物相比略有厚度,且纳米颗粒团聚严重,形成蜂窝状的产物形貌;当表面活性剂为PVA时,所得片状产物横纵比相差不大,较为分散,没有大块的纳米颗粒团聚体;产物经过酸泡后,得到的产物为粒径较小的片状微纳米(K,Na)NbO3晶体,只有少量的纳米颗粒团聚在周边。
通过改变不同的条件,所得产物基本都为片状,化学性能较为稳定,虽然反应的过程中有碱性杂质产生,但经过酸泡后,其产物都为片状,纳米颗粒大量减少。
以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关工作人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。
一种薄片微纳米(K,Na)NbO晶体及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0