









专利摘要
一种使木质素解聚的方法,其包括氧化所述木质素,以提供氧化的木质素,其中β‑O‑4连接的苄型‑OH已被转化为羰基。在存在铵盐或二氧化碳的条件下,用选自由锌、镁、铝和钛或它们的混合物组成的组的金属使氧化的木质素解聚。还描述了用于由木质素制备酚产物的方法和用于切割底物中的β‑O‑4连接的方法。
权利要求
1.一种使木质素解聚的方法,所述方法包括:
氧化所述木质素,以提供氧化的木质素,其中β-O-4连接的苄型-OH已被转化为羰基;并且
在存在铵盐或二氧化碳的条件下,用选自由锌、镁、铝和钛或它们的混合物组成的组的金属使所述氧化的木质素解聚。
2.权利要求1的方法,其中用于所述木质素的氧化剂包括醌。
3.权利要求2的方法,其中,在存在醌和二氧化氮来源的条件下,用分子氧氧化所述木质素。
4.权利要求2或权利要求3的方法,其中工艺作为一锅工艺进行,氧化和解聚两个步骤依次进行。
5.权利要求2-4中任一项的方法,其中所述醌选自由下述各项组成的组:2,3-二氯-5,6-二氰基-1,4-苯醌(DDQ),对-氯苯胺,邻-氯苯胺,苯醌,2-氯蒽醌,和1,4,5,8-四氯蒽醌。
6.权利要求3的方法,或权利要求4或权利要求5在引用权利要求3时的方法,其中所述二氧化氮来源选自由下述各项组成的组:烷基亚硝酸盐,二氧化氮(NO2),亚硝酸盐,硝酸/盐酸混合物和一氧化氮(NO)。
7.权利要求3的方法,其中所述醌以所述木质素的2-30重量%的量使用。
8.权利要求7的方法,其中所述醌以所述木质素的5-20重量%的量使用。
9.权利要求1-8中任一项的方法,其中所述氧化步骤在醇溶剂中或在包含醇溶剂的混合物中进行。
10.权利要求1-8中任一项的方法,其中所述氧化步骤在选自由下述各项组成的组的溶剂中进行:水,甲醇,乙醇,甘油,乙二醇醚和它们的混合物。
11.权利要求9的方法,其中所述氧化步骤在存在选自由下述各项组成的组的共溶剂的存在下进行:烷基醚,烷基腈,羧酸和芳香族溶剂。
12.权利要求9的方法,其中所述氧化步骤在包含下述各项或由下述各项组成或基本上由下述各项组成的溶剂体系中进行:2-甲氧基乙醇,2-甲氧基乙醇和1,2-二甲氧基乙烷的混合物,或2-甲氧基乙醇、1,2-二甲氧基乙烷和水的混合物。
13.权利要求3的方法,或权利要求4-12中任一项在引用权利要求3时的方法,其中所述氧化步骤在20℃-130℃的温度进行。
14.权利要求1的方法,其中所述用于木质素的氧化剂包括下述各项中的一种:2,2,6,6-四甲基哌啶-1-基)氧基(TEMPO)或其衍生物,2-氮杂金刚烷N-氧基,(AZADO),9-氮杂正金刚烷N-氧基,PWC或钒催化剂。
15.任一前述权利要求的方法,其中在所述氧化的木质素的解聚中使用铵盐。
16.权利要求15的方法,其中所述铵盐选自由下述各项组成的组:氯化铵,甲酸铵,乙酸铵,硫酸铵和硫酸氢铵。
17.任一前述权利要求的方法,其中在所述氧化的木质素的解聚中使用锌作为金属。
18.任一前述权利要求的方法,其中所述氧化的木质素的解聚在包含水和醇的溶剂中进行。
19.权利要求18的方法,其中所述氧化的木质素的解聚在包含水和乙二醇醚的溶剂中进行。
20.权利要求19的方法,其中所述氧化的木质素的解聚在包含水和2-甲氧基乙醇的溶剂中进行。
21.权利要求18-20中任一项的方法,其中所述氧化的木质素的解聚在选自由下述各项组成的组的共溶剂的存在下进行:烷基醚,烷基腈,羧酸和芳香族溶剂。
22.权利要求20的方法,其中所述氧化的木质素的解聚在2-甲氧基乙醇∶水的体积比为9∶1-7∶3的水和2-甲氧基乙醇的混合物中进行。
23.任一前述权利要求的方法,其中所述氧化的木质素的解聚在20℃至120℃进行。
24.任一前述权利要求的方法,其还包括从反应混合物中获得根据式I、II或III的任一项的酚:
25.一种使氧化的木质素解聚的方法,其中β-O-4连接的苄型-OH已被转化成羰基,所述方法包括:
在存在铵盐或二氧化碳的条件下,使所述氧化的木质素与选自由下述各项组成的组的金属反应:锌,镁,铝和钛,或它们的混合物。
26.一种制备通式Z的酚产物的方法:
其中R1和R2每次出现时独立地是-H或-OMe,并且R3是-H或-OH;
其中所述方法包括将木质素氧化为氧化的木质素,其中β-O-4连接的苄型-OH已被转化成羰基;并且
在存在铵盐或二氧化碳的条件下,使用选自由锌、镁、铝、钛和它们的混合物组成的组的金属使所述氧化的木质素解聚。
27.权利要求26的方法,其中式Z的酚产物是式I、II或III中的至少一种:
28.一种用于切割底物中的β-O-4连接的方法,所述方法包括:
氧化所述β-O-4连接,以提供氧化的产物,其中β-O-4连接的苄型-OH已被转化成羰基;并且
在存在铵盐或二氧化碳的条件下,用选自由下述各项组成的组的金属使所述氧化的木质素解聚:锌,镁,铝,钛和它们的混合物。
说明书
发明领域
本发明涉及加工木质素以提供有用的小分子。
发明背景
由于全球化石燃料的储备下降并且其价格上涨,由可更新的生物资源制备化学品和燃料的技术已经成为集中发展的焦点。尤其是,认为包含纤维素、半纤维素和木质素的木质纤维素生物质是生物精炼情形中重要的未来资源。尽管用于制备来源于木质纤维素生物质的纤维素成分的乙醇的技术已经投入商业运转,但是利用木质素成分的工艺仍然是有限的。这组成了生物精炼中的重要缺点,在生物精炼中,生物质所有成分的物价稳定对于经济活力是必需的。另外,尽管木质素是纸浆和造纸工业的主要副产物,但是仅有2%的目前产生的木质素得到商业利用。因此,木质素的物价稳定成为还必须需要解决的重要挑战。
木质素构成大部分高等植物的生物质的15-30%和贮能量的多至40%。木质素的异质性质意指单体和结构单位的比例高度取决于植物来源和提取工艺。然而,在几乎所有的木质素中,主要的结构单位是β-O-4(β-芳基醚)连接和较少量的β-β(树脂醇)、β-5(苯基香豆冉(phenylcourmaran))、β-1(1,2-二芳基丙烷)、5-5(联苯基)和4-O-5(二芳基醚)连接。在以下的方案1中提供了木质素聚合结构和芳香结构部分之间的多种连接类型的说明的示意图。芥子醇来源的和松柏醇来源的单位可以分别指定为S和G。
利用木质素制备可生物更新的化学品的主要障碍是该顽固材料向单体的选择性解聚。正是由于这一原因,需要新的化学工艺。已经提议了一些方法。最近,已经提议与第二化学处理联合的木质素选择性氧化作为导致木质素选择性解聚的方法。Rahimi等人(参考文献1)表明,在存在硝酸和盐酸以及4-乙酰胺基-TEMPO的条件下,用分子氧使木质素模型化合物氧化,接着在碱性条件下用过氧化氢切割C-C键的第二步骤。他们提示木质素的可能的应用。Nguyen等人(参考文献2)提议两个步骤的工艺,该工艺也是仅在模型化合物上证明,其中用Bobbitt’s盐([4-AcNH-TEMPO]BF4)氧化是第一步。其可以后接第二步:在存在铱配合物和光的条件下,用胺氢供体和甲酸切割C-O键。
对于发现从木质素获得有用物质的新方法还存在需求。
发明描述
根据第一方面,本发明提供了使木质素解聚的方法,所述方法包括:
氧化木质素,以提供氧化的木质素,其中β-O-4连接的苄型-OH已被转化为羰基;并且
在存在铵盐或二氧化碳的条件下,用选自由锌、镁、铝、钛和它们的混合物组成的组的金属使所述氧化的木质素解聚。
用于木质素的氧化剂可以包括分子氧、醌和二氧化氮来源。备选地,用于木质素的氧化剂可以包括醌。已经发现用醌试剂氧化产生氧化的木质素,其中至少一些β-O-芳基结构部分(β-O-4连接)具有转化为羰基的苄型-OH。
进行相同的β-O-4连接的转化的其他试剂可以用于该方法的氧化步骤。例如,2,2,6,6-四甲基哌啶-1-基)氧基(TEMPO)及其衍生物。例如,4-羟基-TEMP,4-乙酰胺基-TEMPO和其他硝酰基基团,如2-氮杂金刚烷N-氧基(AZADO)或9-氮杂正金刚烷N-氧基。其他的试剂,如Pd/C或钒催化剂,电可以适当地进行该氧化。
任何包含β-O-4连接的木质素都可以用在该工艺中。取决于提取条件,典型的β-O-4连接水平可以是木质素中总连接的约~45-65%。
氧化和解聚步骤可以作为分开的工艺步骤进行。如果这样,那么氧化的木质素可以与初始反应混合物分离,然后进行解聚步骤。
备选地并且便利地,所述方法可以作为一锅工艺进行,氧化和解聚步骤依次进行。已经证明成功地操作了一种便利的一锅工艺,其中氧化步骤在存在醌和二氧化氮来源的条件下使用分子氧,或使用醌作为氧化剂。
氧化步骤可以用2,3-二氯-5,6-二氰基-1,4-苯醌(DDQ)进行。可以使用的其他醌包括对-氯苯胺、邻-氯苯胺、苯醌、2-氯蒽醌和1,4,5,8-四氯蒽醌。已经发现使用醌的氧化步骤至少氧化木质素的β-O-芳基连接。氧化步骤可以使用与木质素大致等重量的氧化剂醌或更多的醌进行。
备选地并且便利地,利用分子氧联合醌(例如DDQ)和二氧化氮(NO2)来源,诸如烷基亚硝酸盐,例如,叔丁基亚硝酸盐。其他二氧化氮来源可以包括NO2本身、亚硝酸盐,诸如NaNO2,硝酸/盐酸混合物或一氧化氮(NO),它们是可商购的。使用这样的氧化系统,醌,诸如DDQ,可以以催化方式使用。例如,以木质素的2-30重量%或甚至木质素的5-20重量%的量。使用所述系统使β-O-4连接中转化成羰基的苄型-OH的比例可以是高的,例如,多至100%,如通过追踪木质素中氧化结构的交叉峰的出现和初始β-O-4连接的交叉峰的消失的2D NMR确定的。更一般地,在醌和二氧化氮来源的存在下涉及醌或分子氧的氧化可以以40%以上的程度在β-O-4连接处氧化木质素,如通过所述2D NMR追踪测量确定的。
氧化步骤便利地在醇溶剂(包含至少一个羟基官能团的溶剂)或溶剂混合物中进行。水也是适当的溶剂。适当的溶剂包括水、甲醇、乙醇、甘油和它们的混合物。乙二醇醚溶剂是便利的,例如,2-甲氧基乙醇。
可以利用共溶剂提高氧化产物的转化。共溶剂可以包括烷基腈,诸如乙腈,羧酸,诸如乙酸,芳香族溶剂,诸如取代的苯。例如,氯代芳族化合物(例如,氯代苯)。有利地,使用烷基醚,例如,乙二醇或丙二醇的二烷基醚,诸如1,2-二甲氧基乙烷。可以使用其他的烷基醚,诸如1,2-二乙氧基乙烷,或环戊基甲醚。这些溶剂的混合物可以与可以包含水的醇溶剂体系一起使用。
当在存在醌和二氧化氮来源的条件下使用分子氧,或使用醌时,氧化步骤可以便利地在环境温度或升高的温度,例如在20-130℃或甚至在60-110℃运行。典型地,反应可以在约80℃运行,比如从70至90℃进行。氧可以在大气压下在反应混合物上方供应。可以通过使氧鼓泡通过反应混合物而改善混合。当使用将氧鼓泡通过反应混合物时,优选地,气氛应该再循环,以避免共氧化剂NO2从该体系中丢失。反应可以在升高的压力下进行,以辅助氧利用。
如果需要,可以在进行解聚步骤之前将氧化的木质素分离。例如,通过添加反溶剂(诸如二乙醚或环戊基甲醚)从包含醇的溶剂体系中沉淀;备选,通过蒸发所使用的溶剂的一些或全部而进行分离。
氧化的木质素(其中至少一些β-O-芳基结构部分(β-O-4连接)具有转化成羰基的苄型-OH)用于解聚步骤。因此,根据第二方面,本发明提供使氧化的木质素解聚的方法,其中β-O-4连接的苄型-OH已被转化成羰基,所述方法包括:
在存在铵盐或二氧化碳的条件下,使氧化的木质素与选自由锌、镁、铝和钛或它们的混合物组成的组的金属反应。
对于根据本发明第一或第二方面所述的解聚,可以使用下述条件:
例如,铵盐可以是氯化铵。金属可以是粉末形式。按成本计,锌可以是优选的。可以使用锌的活性形式,诸如锌-铜偶联物(zinc-copper couple)。当使用活化的锌时,可能不需要存在铵盐或CO2。备用的铵盐包括甲酸铵、乙酸铵、硫酸铵和硫酸氢铵。
作为进一步的备选方案,可以使用酸或酸的混合物,例如,无机酸或有机酸,来激活用于解聚步骤的金属。用于该任务的适当的无机酸可以包括盐酸和硫酸以及它们的混合物。用于本研究的适当的有机酸可以包括羧酸,如甲酸和乙酸以及它们的混合物。
氧化的木质素可以在水性醇溶剂体系中反应从而解聚,例如,在水/乙二醇醚体系中,如水和2-甲氧基乙醇混合物中。可以存在共溶剂,并且可以包括烷基腈,诸如乙腈,羧酸,诸如乙酸,芳香族溶剂,诸如取代的苯。例如,氯代芳族化合物(例如,氯代苯)。有利地,使用烷基醚,例如,乙二醇或丙二醇的二烷基醚,诸如1,2-二甲氧基乙烷。可以使用其他的烷基醚,诸如1,2-二乙氧基乙烷,或环戊基甲醚。这些溶剂的混合物可以与任选地包含水的种醇溶剂或任选地包含水的多种醇溶剂的混合物一起使用。
可以使用9∶1至7∶3的2-甲氧基乙醇:水(体积)的混合物,例如,8∶2的2-甲氧基乙醇:水(体积)的混合物。已经发现所述溶剂体系是用于氧化的木质素的良好的溶剂。作为备选方案,可以使用任选地还包含水的2-甲氧基乙醇和1,2-二甲氧基乙烷的混合物。可以使用在1∶0-1∶2体积比范围内(例如,1∶0-1∶1)的2-甲氧基乙醇:1,2-二甲氧基乙烷。可以使用在反应溶剂总体积的0-50%范围内的水。
解聚步骤可以便利地在环境温度或升高的温度,例如,在20-120℃或甚至在60-110℃运行。典型地,反应可以在约80℃运行,例如从70至90℃进行。反应可以在大气压或升高的压力下进行。
在解聚反应完成后,可以过滤出未反应的金属,用于再利用或丢弃。
然后,可以从反应混合物获得解聚的产物。便利地,利用添加水或加入到水中来沉淀固体,即还没有被转化成低分子量产物的木质素级分。可以将木质素级分过滤出,并且用溶剂清洗,以提取低分子量的产物。类似地,可以用溶剂萃取滤过物,以提取低分子量产物。然后,可以通过任意适当的技术从合并的提取溶剂层中获得低分子量产物,例如,蒸发溶剂,接着对残留物进行色谱(例如,在硅石上),从而提供纯化的化合物。用于萃取目的的适当的溶剂包括乙酸乙酯、叔丁基甲醚和环戊基甲醚。
作为以两个分开的步骤进行本发明第一方面的工艺(即,氧化成氧化的木质素,然后解聚氧化的木质素)的备选方案,该工艺可以作为“一锅”工艺进行。在氧化步骤后,向在醇溶剂体系中包含氧化的木质素的反应混合物中加入水。例如,向使用基于乙二醇醚(例如,2-甲氧基乙醇)的溶剂体系的反应混合物中加入初始溶剂的20体积%的水。然后,如上文讨论那样进行解聚步骤。
在工艺结束时留下的木质素级分可以用于能量产生、碳纤维制备,或可以进一步加工获得有用的产物。例如,作为用于进一步的催化降解的底物,以提供其他小分子产物。例如,其可以进行热解聚,从而产生热解的木质素,其用作燃料资源和/或用于进一步处理,以提供有用的有机化学品(参考文献3)。
已经发现,氧化的木质素的解聚产生酚产物,包括式I、II和III的化合物:
I是3-羟基-1-(4-羟基-3,5-二甲氧基苯基)丙-1-酮;
II是3-羟基-1-(4-羟基-3-甲氧基苯基)丙-1-酮;和
III是1-(4-羟基-3,5-二甲氧基苯基)丙-1-酮。
在下述方案2中示例了木质素的氧化和之后的解聚,其显示部分木质素结构的苄型羟基(A)氧化成相应的羰基(B)。这之后是锌介导的解聚,其涉及在酚醚连接(C-O)处的切割,从而提供酚单体,诸如I。
酚产物可以用作制备有用的有机化学品的中间体。
因此,在第三方面,本发明提供一种制备通式Z的酚产物的方法:
其中R1和R2在每次出现时独立地是-H或-OMe,并且R3是-H或-OH;
其中所述方法包括将木质素氧化为氧化的木质素,其中β-O-4连接的苄型-OH已被转化成羰基;并且
在存在铵盐或二氧化碳的条件下,使用选自由锌、镁、铝、钛和它们的混合物组成的组的金属使所述氧化的木质素解聚。
可以使用所述方法制备至少一种上文所示的根据式I、II或III的化合物,或这些的任意混合物。
第三方面的方法可以使用上文关于本发明的第一和第二方面所提议的溶剂和条件进行。
一种或多种酚产物(例如,式I、II或III的)的分离可以以与关于本发明的第一和第二方面所述相同的方式进行。因此,在反应完成后,可以过滤出未反应的金属,用于再利用或丢弃。
便利地,利用向反应混合物中加入水来沉淀固体,即,还没有转化为酚产物的木质素级分。可以过滤出该木质素级分,并且用溶剂清洗,以提取低分子量酚产物。类似地,可以用溶剂萃取滤过物,以提取低分子量的酚产物。然后,可以通过任意适当的技术从合并的提取溶剂层中获得酚产物,例如,蒸发溶剂,然后对残留物色谱(例如,在硅石上),从而提供纯化的化合物。用于萃取目的的适当的溶剂包括乙酸乙酯。在硅石上的色谱可以使用诸如石油醚:乙酸乙酯(0-55体积%)的溶剂体系。
根据第四方面,本发明提供一种用于切割底物中的β-O-4连接的方法,所述方法包括:
使β-O-4连接氧化,以提供氧化的产物,其中β-O-4连接的苄型-OH已被转化成羰基;并且
在存在铵盐或二氧化碳的条件下,使用选自由锌、镁、铝、钛以及它们的混合物组成的组的金属使氧化的木质素解聚。
第四方面的方法可以使用上文关于本发明的第一和第二方面提出的溶剂和条件进行。
一些优选的实施方案和实验结果的描述
模型化合物研究:
使用DDQ、tBuONO和O2作为氧化体系,使用2-甲氧基乙醇作为溶剂,使甲氧基取代的β-O-4连接的二芳基化合物(1a至1d)和藜芦醇(1e)进行氧化步骤。以下表1每个条目旁所示的高产率获得氧化的产物2a至2e,其突出显示了所用的催化氧化条件的选择性和反应性。
表1
模型聚合物研究
制备结合了等比例的S和G单位的聚合物作为富含β-O-4的硬木木质素的模型,并且使用半定量2D-HSQC NMR实验评估氧化实验的结果。
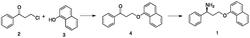
按照下述反应(方案3)制备聚合物:
开始在与单体研究所用的那些相同的条件下(下表2,条目1)处理聚合物。反应的结果表明,尽管仍然进行催化氧化,但是转化低于在获得接近完全转化的情况下的单体系列中所观察到的转化。由此寻求反应条件的进一步优化。将DDQ负荷增加至10mol%显著提高了转化(条目2)。另外发现,加入1,2-二甲氧基乙烷作为非质子(non-protic)共溶剂显著提高转化(条目3和4)。在10mol%DDQ和10mol%tBuONO,氧化达到91%。然而,为了获得几乎完全的氧化,DDQ和tBuONO的负荷达到20mol%(条目6)。在该反应中,共溶剂的有益作用可以部分由在这些条件下DDQ的有效寿命增加解释。有趣的是,注意到G单位似乎比S单位更容易氧化。这一观察与通过苄基阳离子中间体的形成进行的反应相一致,所述苄基阳离子中间体被对-甲氧基取代基共振稳定,但是被间-甲氧基取代基经由诱导的电子收回而去稳定。这意味着,仅被一个间-甲氧基基团取代的G单位比携带两个间-甲氧基基团的S单位更容易氧化。
表2
[a]A=2-甲氧基乙醇,B=2-甲氧基乙醇/1,2-二甲氧基乙烷(2∶3)
[b]转换通过整合2D HSQC NMR中的氧化的和未氧化的结构特有的芳香族交叉峰而确定。误差分析表示这些测量的标准偏差小于<0.05。
木质素实验
在下文所述的使用真正的木质素作为底物的实验中,使用典型的dioxasolv工艺来从桦树锯屑(Betula pendula)中提取木质素。将桦树锯屑在二 烷与2M HCl水溶液的8∶2混合物中使用1∶8的质量:体积比加热至回流1小时。然后,通过过滤分离包含木质素的液体,在减压下部分浓缩,并且在水中沉淀。然后,通过过滤收集沉淀的木质素,溶解在二 烷/水9∶1中,并且通过用二乙醚沉淀进行纯化。分离的木质素富含丁香基(syringyl)并且包含高比例的β-O-4连接,较少量的β-β(树脂醇)和仅有刚刚可检测量的β-5连接(方案1)。在催化DDQ条件下的氧化顺利进行,产生氧化的木质素,其中在2D HSQC NMR谱中可以容易地鉴别需要的α酮β-O-4结构的出现。与分配给α酮结构的交叉峰的出现同时,观察到分配为未氧化的β-O-4结构的交叉峰的完全消失。相对于芳香族区域的总和的整合表明在模型研究中观察到的选择性在木质素本身中保持,原因在于在氧化之前和之后β-O-4交叉峰的相对体积积分几乎是相同的(分别为22.0个质子和23.4个质子/100个芳香族质子)。
木质素氧化的一般过程:
向木质素在2-甲氧基乙醇(14mL/g)或2-甲氧基乙醇/1,2-二甲氧基乙烷(DME)(2∶3,14mL/g)中的溶液中加入DDQ,然后加入tBuONO。将反应混合物放置在O2气氛(气囊)下,并且在80℃搅拌14小时。通过在10体积的Et2O中沉淀和过滤分离氧化的木质素,备选地,将木质素溶液在不经任何进一步的处理的情况下用于下一步骤。
实验结果在下表中给出。
[a]通过在氧化之前和之后对应于丁香基环的NMR谱中S2,6和S’2,6芳香族交叉峰的相对整合确定。与氧化之前和之后木质素中丁香基单位相对应的区域被充分解析,因此,这一比较用作氧化的测量。
锌介导的木质素解聚:
从分离的氧化的木质素:向木质素(600mg)在2-甲氧基乙醇(8.4mL)中的溶液中加入水(2.1mL),然后加入NH4Cl(740mg)和锌粉(900mg)。将反应混合物在80℃加热1小时,冷却并过滤,以去除过滤的锌。然后将反应混合物加入到水(30mL)中,通过添加1M HCl酸化至pH 1(这引起木质素絮凝),然后过滤混合物。将残余的木质素用EtOAc清洗,并且用EtOAc(5x20mL)萃取水性滤液。将有机萃取物和洗液合并,用饱和的NaHCO3、盐水清洗,干燥(MgSO4)并在真空中浓缩。将粗萃取物通过柱色谱(石油醚∶乙酸乙酯(0-55%))纯化,得到3种产物。II(3.5mg,0.58wt%),I(29mg,5wt%)和III(3.4mg,5.7wt%)。
一锅法:按照用于催化氧化的一般步骤,将木质素(2.40g)在2-甲氧基乙醇/1,2-二甲氧基乙烷中处理。在反应时间后,添加水(7mL),然后添加NH4Cl(3.0g)和锌粉(3.60g)。然后,将混合物在80℃加热1小时,冷却,并过滤,以去除过量的锌。然后将反应混合物加入到水(100mL)中,通过添加1M HCl酸化至pH 1(这引起木质素絮凝),然后过滤混合物。将残余的木质素用EtOAc清洗,并且用EtOAc(5x50mL)萃取水性滤液。将有机萃取物和洗液合并,用饱和的NaHCO3、盐水清洗,干燥(经MgSO4)并在真空中浓缩。将粗萃取物通过柱色谱在硅石上用石油醚∶乙酸乙酯(0-55%)纯化,得到3种产物。II(11mg,0.46wt%),I(110mg,4.6wt%)和III(12mg,0.50wt%)。
分离的产物:
3-羟基-1-(4-羟基-3,5-二甲氧基苯基)丙-1-酮(I):白色固体。熔点(M.p.)为109-110℃。光谱数据与在文献中报道的那些一致。
1H NMR(400MHz,CDCl3)6=7.25(s,2H),6.08(s,1H),4.03(t,J=5.4,2H),3.95(s,6H),3.19(t,J=5.4,2H),2.74(s,1H)。
13C NMR(101 MHz,CDCl3)δ199.00,147.0,140.3,128.4,105.6,58.4,56.6(2C),40.0。
3-羟基-1-(4-羟基-3-甲氧基苯基)丙-1-酮(II):浅黄色无定形固体。光谱数据与在文献中报道的那些一致。
1H NMR(500MHz,CDCl3)6=7.58-7.50(m,2H),6.95(d,J=8.1,1H),6.17(s,1H),4.02(t,J=5.3,2H),3.96(s,3H),3.19(t,J=5.3,2H),2.78(s,1H)。
13C NMR(126MHz,CDCl3)δ=199.2,150.9,146.8,129.8,123.8,114.1,109.7,58.5,56.2,39.9。
1-(4-羟基-3,5-二甲氧基苯基)丙-1-酮(III):无色固体。熔点(M.p.)为102-104℃(文献(lit.)109-111℃)。光谱数据与在文献中报道的那些一致。
1H NMR(400MHz,CDCl3)δ=7.27(s,2H),5.92(s,1H),3.96(s,6H),2.97(q,J=7.3,2H),1.23(t,J=7.3,3H)。
13C NMR(101MHz,CDCl3)δ=199.4,146.9,139.6,128.7,105.5(2C),56.6(2C),31.5,8.7。
参考文献
1)A.Rahimi,A.Azarpira,H.Kim,J.Ralph,S.S.Stahl,J.Am.Chem.Soc.2013,135,6415-6418;
2)J.D.Nguyen,B.S.Matsuura,C.R.J.Stephenson,J.Am.Chem.Soc.2013,136,1218-1221。
3)A.Kloekhorst,J.Wildschut,H.J.Heeres,Catal.Sci.Technol.,2014,4,2367-2377。
氧化的木质素的解聚专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0