







专利摘要
本公开涉及一种合成NU‑88分子筛的方法,该方法包括以下步骤:a、将1,3‑二溴丙烷、N‑甲基‑4‑哌啶酮与溶剂混合,在15~60℃预反应0.5~96h,得到预反应产物,所述1,3‑二溴丙烷、N‑甲基‑4‑哌啶酮与溶剂的摩尔比为1:(1.8~3):(1~20);b、将步骤a中得到的预反应产物、有机碱、无机碱、铝源、硅源和水混合,得到待晶化混合物,将所述待晶化混合物进行水热晶化处理,回收固体产物。本公开的方法省去了常规合成NU‑88分子筛模板剂所必经的高成本分离、提纯等繁琐过程,避免了大量的时间消耗、能耗和物耗,降低成本效果明显。
权利要求
1.一种合成NU-88分子筛的方法,其特征在于,该方法包括以下步骤:
a、将1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂混合,在15~60℃预反应0.5~96h,得到预反应产物,所述1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂的摩尔比为1:(1.8~3):(1~20);
b、将步骤a中得到的预反应产物、有机碱、无机碱、铝源、硅源和水混合,得到待晶化混合物,将所述待晶化混合物进行水热晶化处理,回收固体产物。
2.根据权利要求1所述的方法,其中,步骤a中,所述1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂的摩尔比为1:(1.9~2.5):(2~18)。
3.根据权利要求1所述的方法,其中,步骤a中,所述预反应的条件为:15~55℃,时间为1~72h。
4.根据权利要求1所述的方法,其中,步骤a中,所述溶剂为选自水、C1~C6的一元醇、C4~C6的醚、C3~C6的酮、C2~C4的多元醇和C3~C6的酯中的至少一种。
5.根据权利要求1所述的方法,其中,步骤b中,所述待晶化混合物的摩尔组成为SiO
6.根据权利要求1所述的方法,其中,步骤b中,所述有机碱为选自四乙基氢氧化铵、四甲基氢氧化铵和四丙基氢氧化铵中的至少一种。
7.根据权利要求1所述的方法,其中,步骤b中,所述无机碱为选自氢氧化钠、氢氧化钾、氧化钠、氧化钾、碳酸钠和碳酸钾中的至少一种。
8.根据权利要求1所述的方法,其中,步骤b中,所述铝源为选自偏铝酸钠、硝酸铝、硫酸铝、异丙醇铝和醋酸铝中的至少一种。
9.根据权利要求1所述的方法,其中,步骤b中,所述硅源为选自硅胶、硅溶胶、白炭黑和正硅酸乙酯中的至少一种。
10.根据权利要求1所述的方法,其中,步骤b中,所述水热晶化处理包括:先在120-140℃下进行第一阶段水热晶化1-3天,然后再在150-180℃下进行第二阶段热晶化4-14天。
11.根据权利要求1所述的方法,其中,该方法还包括:回收固体产物后进行洗涤、过滤和烘干的步骤。
说明书
技术领域
本公开涉及一种合成NU-88分子筛的方法。
背景技术
NU-88分子筛是由专利USP60277707首次公开的一种新型分子筛。该方法使用双(N-甲基吡咯烷)烷撑基溴盐为模板剂,其中烷撑基的碳数为4~6,在160℃的反应温度下需动态晶化9~22天,才能得到NU-88分子筛。NU-88的分子筛结构尚未得到完全解析,据已有的表征和反应评价结果,推测其可能属于BET家族,具有三维十二元环孔道结构。
Lee S B在文献Journal of catalysis,2003,215:151-170中报道了合成NU-88分子筛的方法。该方法使用的模板剂为1,6-双(N-甲基吡咯烷)己烷溴盐,NU-88分子筛只能在n(SiO2)/n(Al2O3)为60、n(NaOH)/n(SiO2)为0.73、n(H2O)/n(SiO2)为40、n(R)/n(SiO2)为0.15(R为模板剂1,6-双(N-甲基吡咯烷)己烷溴盐)、160℃转动晶化的条件下合成,否则产物为无定形或其它分子筛。
NU-88具有良好的热稳定性和水热稳定性。USP6117307将NU-88应用于加氢催化裂化的催化剂,表现出较高的反应活性剂汽油选择性。NU-88优异的物化性质,使它在石油化工领域可以得到更广泛的应用。
目前,合成NU-88分子筛的方法成本高,制约了它的广泛应用。合成NU-88分子筛的双季铵盐模板剂的成本高是NU-88分子筛成本高的最主要原因之一。
合成双季铵盐模板剂1,6-双(N-甲基吡咯烷)己烷溴盐(1,6-MPH)的方法一般需要两种原料1-甲基吡咯烷(1-MP)和1,6-二溴已烷(1,6-DBH)以一定的比例在适当的溶剂中反应,需采用结晶、多次重结晶方法才能得到较纯净的1,6-MPH,而在结晶、多次重结晶过程中又需冷冻、过滤、有机试剂洗涤、干燥、加适当的有机溶剂溶解、加适当的有机溶剂再析出、过滤、洗涤、干燥等等繁琐的操作,消耗大量的时间,并产生大量的能耗和物耗。
发明内容
本公开的目的是提供一种低成本的合成NU-88分子筛的方法。
为了实现上述目的,本公开提供一种合成NU-88分子筛的方法,该方法包括以下步骤:
a、将1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂混合,在15~60℃预反应0.5~96h,得到预反应产物,所述1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂的摩尔比为1:(1.8~3):(1~20);
b、将步骤a中得到的预反应产物、有机碱、无机碱、铝源、硅源和水混合,得到待晶化混合物,将所述待晶化混合物进行水热晶化处理,回收固体产物。
可选地,步骤a中,所述1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂的摩尔比为1:(1.9~2.5):(2~18)。
可选地,步骤a中,所述预反应的条件为:15~55℃,时间为1~72h。
可选地,步骤a中,所述溶剂为选自水、C1~C6的一元醇、C4~C6的醚、C3~C6的酮、C2~C4的多元醇和C3~C6的酯中的至少一种。
可选地,步骤b中,所述待晶化混合物的摩尔组成为SiO2:Al2O3:M2O:OH-:R:H2O=100:(1.25~10):(2~40):(1~200):(4~30):(600~6000),其中,M2O为碱金属氧化物,R为所述预反应产物,R的摩尔数以所述1,3-二溴丙烷的摩尔数计。
可选地,步骤b中,所述有机碱为选自四乙基氢氧化铵、四甲基氢氧化铵和四丙基氢氧化铵中的至少一种。
可选地,步骤b中,所述无机碱为选自氢氧化钠、氢氧化钾、氧化钠、氧化钾、碳酸钠和碳酸钾中的至少一种。
可选地,步骤b中,所述铝源为选自偏铝酸钠、硝酸铝、硫酸铝、异丙醇铝和醋酸铝中的至少一种。
可选地,步骤b中,所述硅源为选自硅胶、硅溶胶、白炭黑和正硅酸乙酯中的至少一种。
可选地,步骤b中,所述水热晶化处理包括:先在120-140℃下进行第一阶段水热晶化1-3天,然后再在150-180℃下进行第二阶段热晶化4-14天。
可选地,该方法还包括:回收固体产物后进行洗涤、过滤和烘干的步骤。
通过上述技术方案,本公开先将合成模板剂的原料预反应一段时间,然后再将其与合成分子筛的其它原料以一定的比例混合,水热晶化合成了NU-88分子筛。本公开的方法省去了常规合成NU-88分子筛模板剂所必经的高成本分离、提纯等繁琐过程,避免了大量的时间消耗、能耗和物耗,降低成本效果明显;此外,也克服了现有技术中将合成模板剂的原料与合成分子筛的其它原料直接混合晶化所带来的原料反应不完全、需回收利用的弊端,避免了由此产生的反应原料的浪费和无效消耗。采用本公开的方法所合成的NU-88分子筛的结晶度较高。
本公开的其他特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
附图是用来提供对本公开的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本公开,但并不构成对本公开的限制。在附图中:
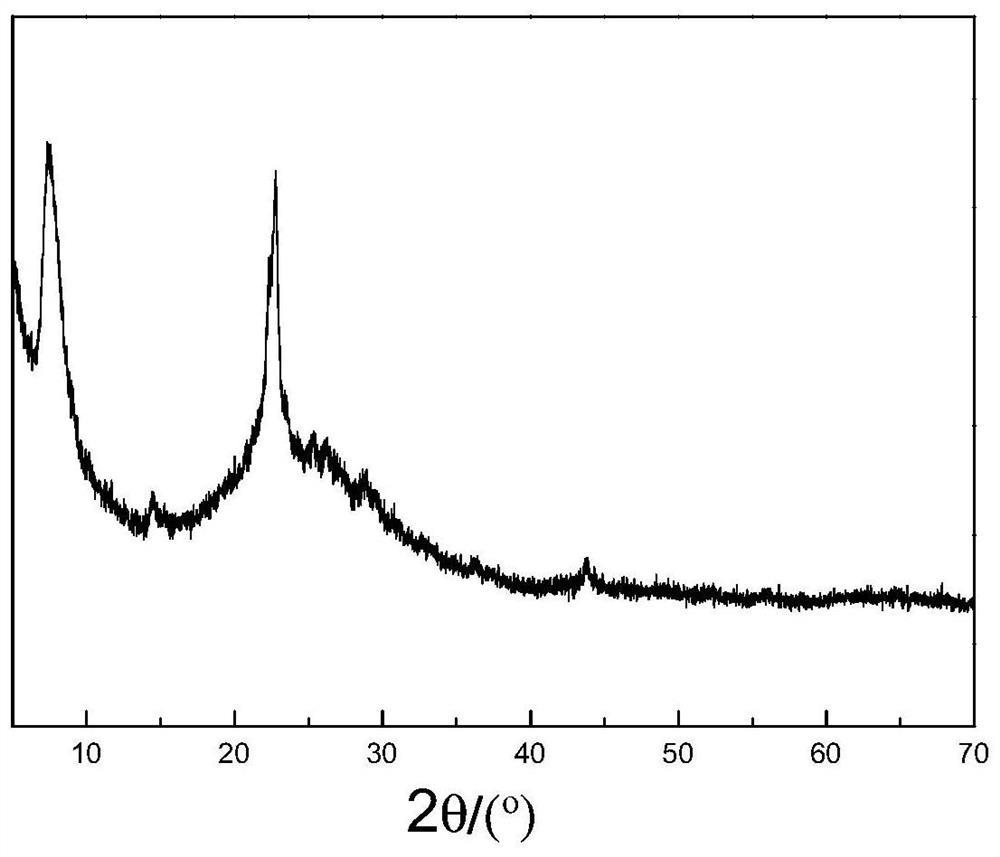
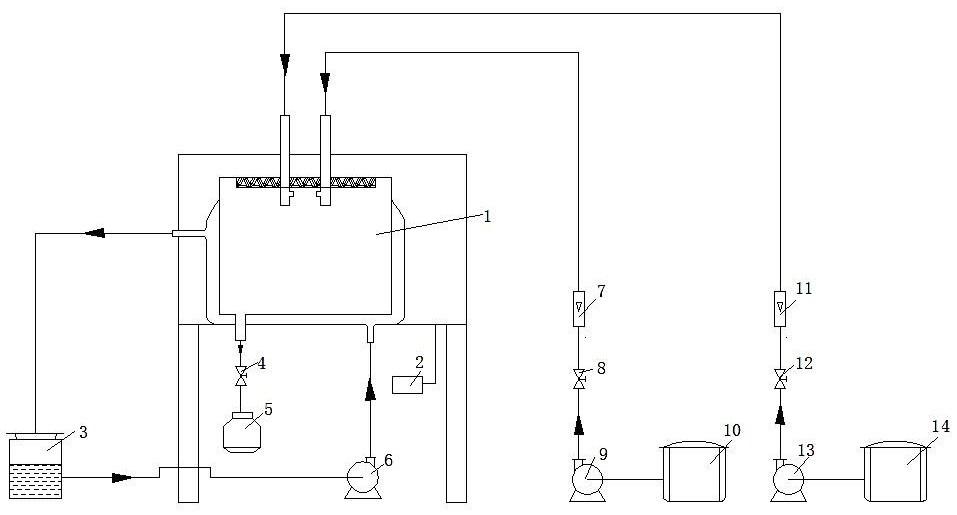
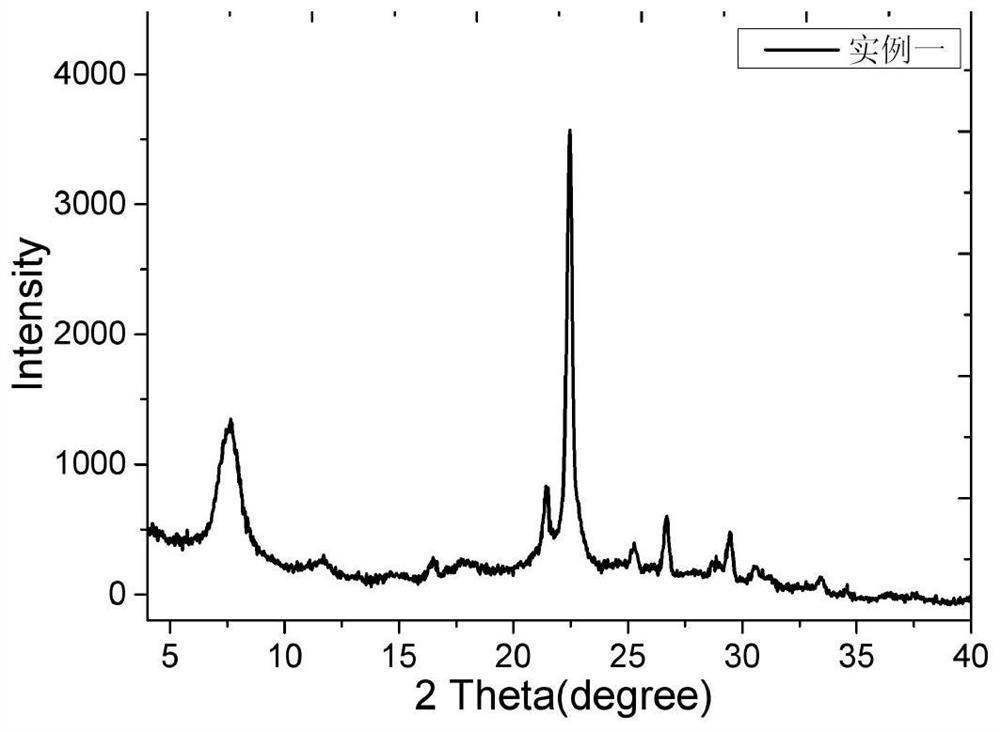
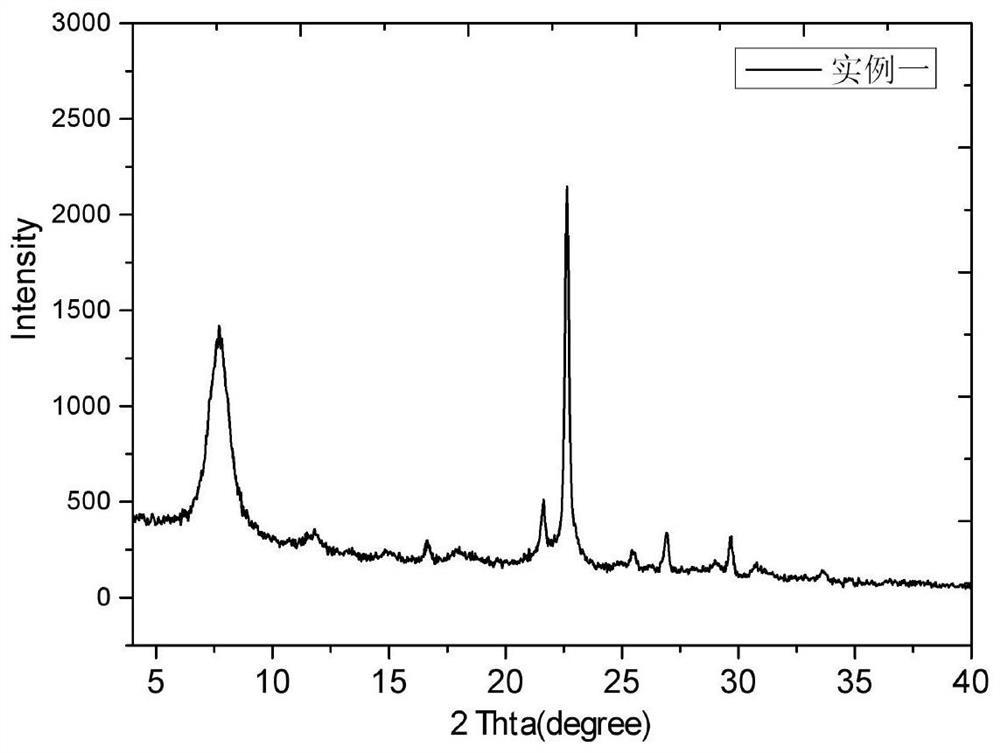
图1是实施例1中合成的NU-88分子筛的X射线衍射谱图。
具体实施方式
以下结合附图对本公开的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本公开,并不用于限制本公开。
本公开提供一种合成NU-88分子筛的方法,该方法包括以下步骤:
a、将1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂混合,在15~60℃预反应0.5~96h,得到预反应产物,所述1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂的摩尔比为1:(1.8~3):(1~20);
b、将步骤a中得到的预反应产物、有机碱、无机碱、铝源、硅源和水混合,得到待晶化混合物,将所述待晶化混合物进行水热晶化处理,回收固体产物。
本公开先将合成模板剂的原料预反应一段时间,然后再将其与合成分子筛的其它原料以一定的比例混合,水热晶化合成了NU-88分子筛。本公开的方法省去了常规合成NU-88分子筛模板剂所必经的高成本分离、提纯等繁琐过程,避免了大量的时间消耗、能耗和物耗,降低成本效果明显;此外,也克服了现有技术中将合成模板剂的原料与合成分子筛的其它原料直接混合晶化所带来的原料反应不完全、需回收利用的弊端,避免了由此产生的反应原料的浪费和无效消耗。采用本公开的方法所合成的NU-88分子筛的结晶度较高。
根据本公开,所述1,3-二溴丙烷的英文名为1,3-Dibromopropane,CAS号为109-64-8,结构式为:
根据本公开,所述N-甲基-4-哌啶酮的英文名为1-Methyl-4-piperidone,CAS号为1445-73-4,结构式为:
根据本公开,步骤a中,所述1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂的摩尔比优选为1:(1.9~2.5):(2~18),进一步优选为1:(2~2.3):(3~16)。
根据本公开,步骤a中,1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂的混合方式可以为本领域常规的,优选地,可以先将N-甲基-4-哌啶酮与溶剂混合,再将1,3-二溴丙烷滴加到上述所得的混合液中,所述滴加的速度可以为1~60滴/秒。所述预反应可以在密闭反应釜或带回流装置的反应釜中进行。所述预反应的条件优选可以为:15~55℃,时间为1~72h。为了获得理想的效果,所述混合以及所述预反应可以在搅拌的条件下进行。
根据本公开,步骤a中,所述溶剂可以为常见的有机溶剂和水,只要满足其能够与1,3-二溴丙烷和/或N-甲基-4-哌啶酮互溶即可,例如,所述溶剂可以为选自水、C1~C6的一元醇、C4~C6的醚、C3~C6的酮、C2~C4的多元醇和C3~C6的酯中的至少一种。优选地,所述溶剂为选自水、C1~C4的一元醇、C4~C5的醚、C3~C4的酮、C2~C3的多元醇和C3~C4的酯中的至少一种;具体地,所述溶剂可以为水、甲醇、乙醇、乙醚、丙酮等。
根据本公开,步骤b中,所述预反应产物、有机碱、无机碱、铝源、硅源和水的混合方式可以为本领域常规的,优选地,可以先将所述预反应产物、有机碱、无机碱和铝源溶于水中,得到混合溶液;再在搅拌条件下,将所述混合溶液与硅源接触,得到待晶化混合物。所述待晶化混合物的摩尔组成可以为SiO2:Al2O3:M2O:OH-:R:H2O=100:(1.25~10):(2~40):(1~200):(4~30):(600~6000),优选为100:(1.25~8):(5~30):(10~150):(5~20):(600~5000),其中,M2O为碱金属氧化物,R为所述预反应产物,R的摩尔数以所述1,3-二溴丙烷的摩尔数计。
根据本公开,步骤b中,所述有机碱、无机碱、铝源、硅源可以为用于合成NU-88分子筛的常规种类。例如,所述有机碱可以为选自四乙基氢氧化铵、四甲基氢氧化铵和四丙基氢氧化铵中的至少一种。所述无机碱可以为选自氢氧化钠、氢氧化钾、氧化钠、氧化钾、碳酸钠和碳酸钾中的至少一种。所述铝源可以为选自偏铝酸钠、硝酸铝、硫酸铝、异丙醇铝和醋酸铝中的至少一种。所述硅源为可以为选自硅胶、硅溶胶、白炭黑和正硅酸乙酯中的至少一种。
根据本公开,步骤b中,所述水热晶化处理可以包括:先在120-140℃下进行第一阶段水热晶化1-3天,然后再在150-180℃下进行第二阶段热晶化4-14天。为了获得理想的效果,所述水热晶化可以在搅拌的条件下进行。
根据本公开,该方法还可以包括:回收固体产物后进行洗涤、过滤和烘干的步骤。其中,所述洗涤、过滤和烘干为合成分子筛的常规步骤,本公开对其条件没有特殊的限制。例如,所述烘干的条件可以为:温度为80~120℃,时间为8~24h。
下面通过实施例对本公开做进一步的说明,但并不因此而限制本公开的内容。
实施例和对比例中“相对结晶度”的相对概念和计算方式为:以对比例1合成的分子筛产品Z0在XRD谱图的2θ区间为21.0-27.5°内的峰面积之和为基准,以百分数计为100%;以下实施例和对比例中合成的产品在XRD谱图上2θ区间为21.0-27.5°内的峰面积之和与Z0的XRD谱图的峰面积之和的比值(以百分数计)为相应产品的相对结晶度值(R.C.%)。
实施例和对比例中,XRD分析采用日本理学D/MAX-ⅢA型衍射仪,测试条件:Cu靶,Kα辐射,Ni滤波片,管电压35kV,管电流35mA,扫描范围2θ为4-55°。
实施例和对比例中,所用到的各种试剂的规格和来源如下:
NaOH、无水乙醇、甲醇、丙酮、乙醚,均为分析纯,北京化工厂生产;
1,6-双(N-甲基吡咯烷)已烷溴盐(1,6-MPH)水溶液,固含量58重量%,由广州大有精细化工厂生产;
四乙基氢氧化铵溶液(TEAOH),固含量35重量%,由广州大有精细化工厂生产;
1,3-双(N-甲基-4-哌啶酮)丙撑基溴盐(MPOP)水溶液,固含量58重量%,按照CN106276951A公开的方法制备得到;
N-甲基-4-哌啶酮(MPO),>98.0重量%,东京化成工业株会社;
1,3-二溴丙烷(1,3-DBP),>98.0%,东京化成工业株会社;
白炭黑,SiO2含量为97重量%,株洲兴隆公司;
NaAlO2溶液,Al2O3含量为13.64重量%,Na2O含量为20.2重量%,中石化股份有限公司长岭催化剂分公司生产。
实施例和对比例中,合成分子筛的成本估算方法为:合成分子筛的所有原料的价格总和与所得产物的重量比。
对比例1
本对比例用于说明专利USP6027707实例1中公开的合成NU-88分子筛的方法,具体步骤如下:
将14.8g 30重量%NaOH溶液与16.115gNaAlO2溶液溶入适量去离子水中,在搅拌的条件下加入40g白炭黑,再加入1,6-双(N-甲基吡咯烷)已烷溴盐(1,6-MPH)水溶液77.08g,混合均匀,制成胶体状待晶化混合物,其摩尔组成为5(1,6-MPH):5Na2O:1Al2O3:30SiO2:1200H2O。将所得转移至1L不锈钢晶化釜中,于160℃下搅拌水热晶化13天。然后停止晶化反应,产物经洗涤、过滤,110℃烘干8h即得到分子筛原粉Z0。
将分子筛原粉Z0的XRD测定结果与专利USP6027707公开的NU-88分子筛XRD谱图对比,确定Z0为NU-88分子筛。设定分子筛原粉Z0的相对结晶度为100%。
对比例2
本对比例用于说明专利CN 106276951A中公开的合成NU-88分子筛的方法,具体步骤如下:
将12.086g NaAlO2溶液、27.229g四乙基氢氧化铵溶液、10.19g 30重量%NaOH溶液、71.58g模板剂1,3-双(N-甲基-4-哌啶酮)丙撑基溴盐溶入适量去离子水中,搅拌条件下,加入白炭黑40g,混合均匀,制成胶体。胶体的摩尔组成为:6(1,3-MPOP):4(TEAOH):4.8Na2O:1Al2O3:40SiO2:1200H2O。将制得的胶体转移至1L不锈钢晶化釜中,于120℃下搅拌晶化2天,再升温至160℃下搅拌晶化10天后,停止晶化反应,产物经洗涤、过滤,110℃烘干8h即得到分子筛原粉Z1,通过XRD分析方法进行测定,与USP6027707公开的方法制备的NU-88分子筛的XRD谱图相比较后,确定Z1为NU-88分子筛,计算其相对结晶度、分子筛收率和合成成本列于表1。
对比例3
本对比例用于说明采用合成模板剂的前驱物N-甲基-4-哌啶酮(MPO)和1,3-二溴丙烷(1,3-DBP)替代有机模板剂1,3-双(N-甲基-4-哌啶酮)丙撑基溴盐(MPOP)直接合成TNU-9分子筛的方法。
按照对比例2的方法合成NU-88分子筛,区别在于,采用33.6g N-甲基-4-哌啶酮(MPO)和20g1,3-二溴丙烷(1,3-DBP)替代1,3-双(N-甲基-4-哌啶酮)丙撑基溴盐(MPOP),N-甲基-4-哌啶酮和1,3-二溴丙烷的摩尔比为3:1。合成得到分子筛原粉Z2。
对分子筛原粉Z2进行XRD测试,并将测定结果与专利USP6027707公开的NU-88分子筛XRD谱图对比,经对比可确定Z2为NU-88分子筛,计算其相对结晶度、分子筛收率和合成成本列于表1。
实施例1-7用于说明本公开的合成NU-88分子筛的方法。
实施例1
在搅拌条件下,将25.2gN-甲基-4-哌啶酮和23mL无水乙醇混合,再将20g1,3-二溴丙烷以5滴/秒的速度滴加到上述混合液中,在50℃下预反应4h,得预反应产物A1。1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂乙醇的摩尔比为1:2.25:4.07。
将预反应产物A1、12.086g NaAlO2溶液、27.229g四乙基氢氧化铵溶液(TEAOH)、10.19g 30重量%NaOH溶液溶于适量去离子水中,混合均匀,在搅拌的条件下,缓慢加入40g白炭黑,制成乳白色胶体状待晶化混合物,其摩尔组成为SiO2:Al2O3:Na2O:TEAOH:A1:H2O=100:3.3:12:10:15:4000,继续搅拌1h,转移至带机械搅拌的1L高压反应釜中,于120℃下搅拌晶化2天,再升温至160℃下搅拌晶化10天后,停止晶化反应,产物经洗涤、过滤后,110℃烘干8h即得到分子筛原粉B1。
对分子筛原粉B1进行XRD测试,谱图如图1所示。将所得XRD谱图与专利USP6027707公中公开的NU-88分子筛的XRD谱图相比较后可确定B1为NU-88分子筛,计算其相对结晶度和合成成本列于表1。
实施例2
按照实施例1的方法合成NU-88分子筛,区别在于,在搅拌条件下,将22.4gN-甲基-4-哌啶酮和32mL乙醚混合,再将20g1,3-二溴丙烷以4滴/秒的速度滴加到上述混合液中,在35℃下预反应12h,得预反应产物A2。1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂乙醚的摩尔比为1:2:3.18。以预反应产物A2替代A1,得到分子筛原粉B2,其XRD谱图与实施例1一致,计算其相对结晶度和合成成本列于表1。
实施例3
按照实施例1的方法合成NU-88分子筛,区别在于,在搅拌条件下,将23.52gN-甲基-4-哌啶酮与27mL去离子水混合,再将20g1,3-二溴丙烷以5滴/秒的速度滴加到上述混合液中,在55℃下预反应48h,得预反应产物A3。1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂水的摩尔比为1:2.1:15.5。以预反应产物A3替代A1,得到分子筛原粉B3,其XRD谱图与实施例1一致,计算其相对结晶度和合成成本列于表1。
实施例4
按照实施例1的方法合成NU-88分子筛,区别在于,在搅拌条件下,将28gN-甲基-4-哌啶酮与70mL甲醇混合,再将20g1,3-二溴丙烷以20滴/秒的速度滴加到上述混合液中,在40℃下预反应20h,得预反应产物A4。1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂甲醇的摩尔比为1:2.5:17.8。以预反应产物A4替代A1,得到分子筛原粉B4,其XRD谱图与实施例1一致,计算其相对结晶度和合成成本列于表1。
实施例5
按照实施例1的方法合成NU-88分子筛,区别在于,在搅拌条件下,将21.28gN-甲基-4-哌啶酮与38mL丙酮混合,再将20g1,3-二溴丙烷以10滴/秒的速度滴加到上述混合液中,在40℃下预反应6h,得预反应产物A5。1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂丙酮的摩尔比为1:1.9:5.32。以预反应产物A5替代A1,得到分子筛原粉B5,其XRD谱图与实施例1一致,计算其相对结晶度和合成成本列于表1。
实施例6
按照实施例1的方法合成NU-88分子筛,区别在于,在搅拌条件下,将110mL乙醇与20g1,3-二溴丙烷的混合液以30滴/秒的速度滴加到30.2gN-甲基-4-哌啶酮中,50℃下预反应10h,得预反应产物A6。1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂乙醇的摩尔比为1:2.7:19.46。以预反应产物A6替代A1,得到分子筛原粉B6,其XRD谱图与实施例1一致,计算其相对结晶度和合成成本列于表1。
实施例7
按照实施例1的方法合成NU-88分子筛,区别在于,在搅拌条件下,将11mL乙醇与20g1,3-二溴丙烷的混合液以2滴/秒的速度滴加到20.16gN-甲基-4-哌啶酮中,75℃下预反应60h,得预反应产物A7。1,3-二溴丙烷、N-甲基-4-哌啶酮与溶剂乙醇的摩尔比为1:1.8:1.94。以预反应产物A7替代A1,得到分子筛原粉B7,其XRD谱图与实施例1一致,计算其相对结晶度和合成成本列于表1。
表1
由表1可见,本公开的方法能够显著降低NU-88分子筛的合成成本,并且所制备的NU-88分子筛具有较高的相对结晶度。
以上结合附图详细描述了本公开的优选实施方式,但是,本公开并不限于上述实施方式中的具体细节,在本公开的技术构思范围内,可以对本公开的技术方案进行多种简单变型,这些简单变型均属于本公开的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本公开对各种可能的组合方式不再另行说明。
此外,本公开的各种不同的实施方式之间也可以进行任意组合,只要其不违背本公开的思想,其同样应当视为本公开所公开的内容。
合成NU-88分子筛的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



动态评分
0.0