









专利摘要
本发明提供了一种表面负载颗粒的棒状MoO3/ZnMoO4材料的合成方法,属于功能性无机材料制备领域,包括以下步骤:将浓HNO3、H2O和异丙醇混合均匀获得混合溶剂,然后加入H24Mo7N6O24.4H2O和三甲基十八烷基氯化铵搅拌均匀,得到混合溶液;然后进行溶剂热反应得到MoO3;以甲醇和丙三醇为溶剂,将含有2‑甲基咪唑和苯并咪唑的溶液B加入到含有MoO3、PVP、1‑乙烯基‑2‑吡咯烷酮和可溶性锌盐的溶液A中,搅拌后干燥得到前驱体;再进行热处理,得到最终产物。产物的形状为表面负载颗粒的棒状,棒的直径为0.1‑0.7μm,长径比为15‑25:1,颗粒粒径为0.03‑0.3μm。本发明所采用的化学试剂价格低廉,实验参数易于控制,所得产品形貌特殊稳定,产量大,有利于MoO3/ZnMoO4规模化生产。
权利要求
1.一种表面负载颗粒的棒状MoO
(1)将浓HNO
(2)将混合溶液进行溶剂热反应,经过洗涤、干燥后得到MoO
(3)将MoO
(4)将2-甲基咪唑和苯并咪唑加入到甲醇和丙三醇的混合溶液中,搅拌均匀,得到溶液B;
(5)将溶液B加入到溶液A中,搅拌一段时间,经过洗涤、干燥后,得到前驱体;
(6)将前驱体进行热处理,得到最终产物;
步骤(1)中,所述混合溶剂中浓HNO
所述MoO
步骤(1)中,所述H
步骤(2)中,溶剂热反应温度为160-200℃,反应时间为15-24h;
步骤(6)中,热处理温度为450-600℃,升温速度为1-3 ℃/min,保温时间为1.5-8 h。
2.根据权利要求1所述的合成方法,其特征在于,步骤(1)中,浓HNO
3.根据权利要求1所述的合成方法,其特征在于,PVP的摩尔数按单体计算,其单体摩尔质量为111;PVP的分子量小于100万。
4.根据权利要求1所述的合成方法,其特征在于,步骤(1)中,所述H
步骤(3)中,MoO
5.根据权利要求1所述的合成方法,其特征在于,步骤(5)中,搅拌时间为1-5 h。
6.根据权利要求1所述的合成方法,其特征在于,步骤(3)中,可溶性锌盐为锌的硝酸盐。
7.根据权利要求1所述的合成方法,其特征在于,步骤(3)、(4)中,甲醇:丙三醇的体积比为1:0.005-0.015。
8.根据权利要求1所述的合成方法,其特征在于,步骤(6)中,热处理在空气气氛下进行。
9.一种如权利要求1-8任一合成方法获得的MoO
说明书
技术领域
本发明属于功能性无机材料制备领域,具体涉及一种表面负载颗粒的棒状MoO3/ZnMoO4结构的合成方法。
背景技术
环境污染的检测与防治是当今社会发展面临的重要课题,尤其是气体污染问题已成为制约我国可持续发展的障碍因素。探索便携性好、灵敏度高且成本较低的气敏传感器材料受到了国内外学者的普遍关注。半导体金属氧化物能够利用电阻的变化检测待测气体的成分和浓度,具有恢复性好、使用寿命长、稳定性高、灵敏度好等优点,在气敏材料领域应用前景广泛。
MoO3是一种n型半导体氧化物(Eg=3.1 eV),在催化剂、气敏传感器、电化学等领域具有广泛应用。MoO3具有h-MoO3和α-MoO3两种常见晶型结构,采用不同的合成方法能够有效调控MoO3产物的微观形貌与物相结构,显示出独特的气敏性质。例如,“Wang L, Gao P,Bao D, et al., Synthesis of Crystalline/Amorphous Core/Shell MoO3 Compositesthrough a Controlled Dehydration Route and Their Enhanced Ethanol SensingProperties, Crystal Growth & Design, 2014, 14(14):569-575.”选择钼氧化物作为原料,通过溶剂热反应,合成了分散均匀的MoO3一维结构,发现其对乙醇具有良好的气敏响应;“Sui L, Zhang X, Cheng X, et al., Au-Loaded Hierarchical MoO3 HollowSpheres with Enhanced Gas-Sensing Performance for the Detection of BTX(Benzene, Toluene, And Xylene) And the Sensing Mechanism, ACS AppliedMaterials & Interfaces, 2016, 9(2):1661.”结合溶剂热法和煅烧法合成了形貌可控的MoO3中空球,对苯、甲苯、二甲苯等显示出良好的气体灵敏性。
其中,MoO3一维微纳米材料具有比表面积大、表面活性高、电子传输过程易于调控等特点,在气敏传感领域可作为物化性能优异的基体材料。最近几年,MoO3基一维复合气敏材料的研发已成为该领域的研究热点。然而,国内外尚未发现采用溶剂热法、室温搅拌法与热处理法相结合的方式制备表面负载颗粒的MoO3/ZnMoO4棒状结构的相关报道。通过设计新颖的反应体系,控制晶体生长动力学过程,进而调控MoO3/ZnMoO4棒状结构的物相组成与微观形貌,具有重要的科学和实践意义。
发明内容
针对现有技术中存在的问题,本发明提供了一种表面负载颗粒的MoO3/ZnMoO4复合材料棒状结构的合成方法,该方法实验参数易于控制,产物形貌重复性好,产量大,适合大规模生产。
为实现上述目的,本发明采用如下技术方案。
一种表面负载颗粒的棒状MoO3/ZnMoO4材料的合成方法,包括以下步骤:
(1)将浓HNO3、H2O和异丙醇混合均匀获得混合溶剂,然后加入H24Mo7N6O24
(2)将混合溶液进行溶剂热反应,经过洗涤、干燥后得到MoO3;
(3)将MoO3加入到甲醇和丙三醇的混合溶液中,经过超声后,加入PVP、1-乙烯基-2-吡咯烷酮和可溶性锌盐,搅拌均匀,得到溶液A;
(4)将2-甲基咪唑和苯并咪唑加入到甲醇和丙三醇的混合溶液中,搅拌均匀,得到溶液B;
(5)将溶液B加入到溶液A中,搅拌一段时间,经过洗涤、干燥后,得到前驱体;
(6)将前驱体进行热处理,得到最终产物。
步骤(1)中,所述浓HNO3、H2O和异丙醇的体积比为1:3-7:0.01-0.05;浓HNO3的质量分数浓度为69 %。
步骤(1)中,所述H24Mo7N6O24
步骤(2)中,溶剂热反应温度为160-200℃,反应时间为15-24h。
步骤(3)中,可溶性锌盐为锌的硝酸盐。
步骤(3)中,MoO3的在甲醇和丙三醇混合溶剂中的浓度为0.08-0.15 mol/L。
步骤(3)、(4)中,甲醇:丙三醇的体积比为1:0.005-0.015。所述MoO3:PVP:1-乙烯基-2-吡咯烷酮:可溶性锌盐:2-甲基咪唑:苯并咪唑的摩尔比为1:0.5-1:0.005-0.015:0.005-0.25:0.05-1.5:0.003-0.10;其中,PVP的摩尔数按单体计算,其单体摩尔质量为111。优选的,PVP的分子量小于100万。
步骤(5)中,搅拌时间为1-5 h。
步骤(6)中,热处理温度为450-600℃,升温速度为1-3 ℃/min,保温时间为1.5-8h。优选的,热处理在空气气氛下进行。
上述制备方法获得的MoO3/ZnMoO4复合材料,形状为表面负载颗粒的棒状,表面负载颗粒为ZnMoO4;棒的直径为0.1-0.7 μm,长径比为15-25:1,颗粒粒径为0.03-0.3 μm。
本发明的制备方法中,在溶剂热和室温搅拌过程中都引入了适量的功能试剂。例如,三甲基十八烷基氯化铵在溶剂热反应中能够提高溶液的粘度,降低晶核长大的速度,且有效吸附于MoO3晶核的晶面,控制了晶体生长习性,形成长径比可调的棒状结构;PVP和1-乙烯基-2-吡咯烷酮在室温搅拌过程中调节了混合溶剂的粘性,能够吸附于MoO3棒状结构表面,改善表面的结合能,有利于Zn
本发明具有以下优点:
本发明选择浓HNO3、H2O和异丙醇作为溶剂热反应时的混合溶剂,以H24Mo7N6O24
本发明设计了新颖的反应体系,采用多种表面活性剂,通过多步反应有效控制了晶体生长速度和物相结构,得到了分散性好、尺寸分布范围窄的前驱体,并通过热处理方法,最终得到了一种表面负载颗粒的MoO3/ZnMoO4棒状结构,与国内外报道的MoO3/ZnMoO4复合材料的合成机理具有本质的区别。本发明所采用的化学试剂价格低廉,实验参数易于控制,所得产品形貌特殊、稳定,产量大,有利于MoO3/ZnMoO4复合材料的规模化生产。
附图说明
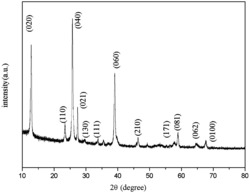
图1为实施例1合成的MoO3材料的X射线衍射(XRD)图谱;
图2为实施例1合成的MoO3/ZnMoO4复合材料的X射线衍射(XRD)图谱;
图3为实施例1合成的MoO3/ZnMoO4复合材料的扫描电镜(SEM)图片。
具体实施方式
下面结合实施例和附图对本发明做进一步说明,但本发明不受下述实施例的限制。
下述实施例中,所用PVP的分子量为40000,PVP的摩尔数按单体计算,其单体摩尔质量为111。
实施例1
(1)将4 mL的HNO3、20 mL的H2O和0.15 mL异丙醇混合均匀,然后加入0.5000 g的H24Mo7N6O24
(2)将得到混合溶液转移至反应釜中,密封后放入真空干燥箱,设置参数为:200℃,保温20 h,然后离心、洗涤、干燥得到MoO3;
(3)将得到的0.25 g的MoO3加入到15 mL甲醇和0.15 mL丙三醇的溶液中,经过超声后,再加入0.1500 g的PVP、0.0017 g的1-乙烯基-2-吡咯烷酮和0.0148 g的Zn (NO3)2
(4)将0.0164 g的2-甲基咪唑、0.0010 g的苯并咪唑溶解到15 mL甲醇和0.15 mL丙三醇的混合溶液中,搅拌均匀,得到B溶液;
(5)将B溶液加入到A溶液中,搅拌2 h后,经过洗涤、干燥后,得到前驱体;
(6)将前驱体置于马弗炉中,在空气气氛下按照1 ℃/min的升温速度由室温升至450 ℃,保温2 h,样品随炉冷却后得到产物。
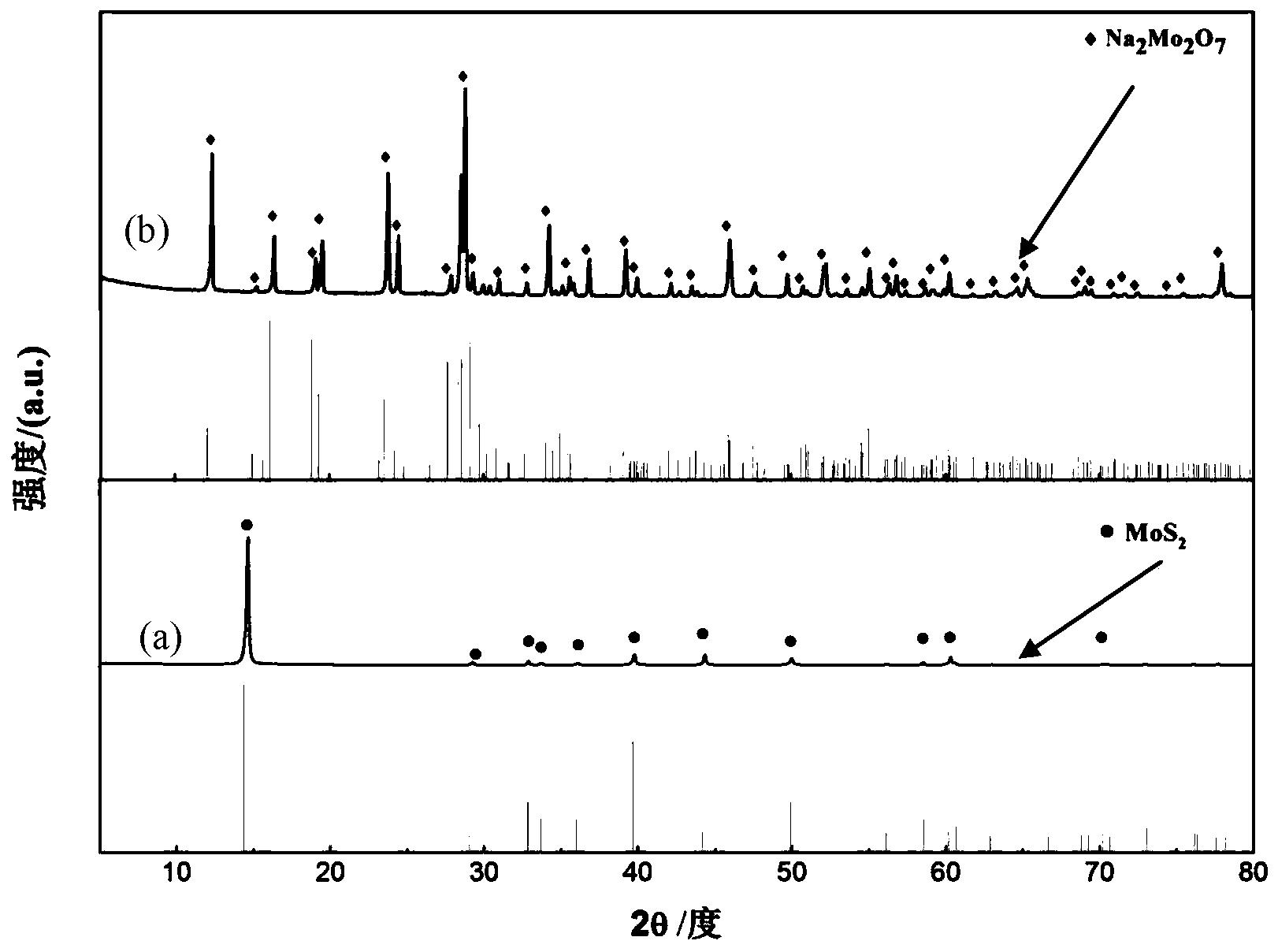
步骤(1)中合成的产物的XRD结果如图1所示,从图中可以看出,所有的衍射峰均与标准XRD卡片(65-2421)保持一致,证明所得产物为MoO3晶相。热处理后得到的最终产物的XRD结果如图2所示,所有的衍射峰均与标准XRD卡片(35-0765和65-2421)保持一致,证明所得产物为MoO3/ZnMoO4复合材料,且结晶性好。其中,◆标注的是ZnMoO4的衍射峰,●标注的是MoO3的衍射峰;产物的SEM照片如图3所示,从图中可以看出,本发明得到的产物为表面负载颗粒的MoO3/ZnMoO4棒状结构,棒的直径为0.35-0.42 μm,长径比为15-16:1,颗粒粒径为0.16-0.18 μm。
实施例2
(1)将3 mL的HNO3、18 mL的 H2O和0.10 mL异丙醇混合均匀,然后加入0.4942 g的H24Mo7N6O24
(2)将得到混合溶液转移至反应釜中,密封后放入真空干燥箱,设置参数为:190℃,保温21 h,然后离心、洗涤、干燥,得到MoO3;
(3)将得到的0.2500 g的MoO3加入到14 mL甲醇和0.10 mL丙三醇的溶液中,经过超声后,再加入0.1600 g的PVP、0.0012 g的1-乙烯基-2-吡咯烷酮和0.0030 g的Zn (NO3)2
(4)将0.0083 g的2-甲基咪唑、0.0007 g的苯并咪唑溶解到14 mL甲醇和0.10 mL丙三醇的混合溶液中,搅拌均匀,得到B溶液;
(5)将B溶液加入到A溶液中,搅拌1.5 h后,经过洗涤、干燥后,得到前驱体;
(6)将前驱体置于马弗炉中,在空气气氛下按照1 ℃/min的升温速度由室温升至480 ℃,保温1.5 h,样品冷却后得到表面负载颗粒的MoO3/ZnMoO4棒状结构,棒的直径为0.25-0.35 μm,长径比为18-19:1,颗粒粒径为0.07-0.13 μm。
实施例3
(1)将5 mL的HNO3、23 mL的H2O和0.17 mL异丙醇混合均匀,然后加入0.5017 g的H24Mo7N6O24
(2)将得到混合溶液转移至反应釜中,密封后放入真空干燥箱,设置参数为:185℃,保温22 h,然后离心、洗涤、干燥得到MoO3;
(3)将得到的0.25 g的MoO3加入到16 mL甲醇和0.18 mL丙三醇的溶液中,经过超声后,再加入0.1400 g的PVP、0.0020 g的1-乙烯基-2-吡咯烷酮和0.0446 g的Zn (NO3)2
(4)将0.0985 g的2-甲基咪唑、0.0096 g的苯并咪唑溶解到16 mL甲醇和0.18 mL丙三醇的混合溶液中,搅拌均匀,得到B溶液;
(5)将B溶液加入到A溶液中,搅拌3 h后,经过洗涤、干燥后,得到前驱体;
(6)将前驱体置于马弗炉中,在空气气氛下按照2℃/min的升温速度由室温升至480 ℃,保温7 h,样品冷却后得到表面负载颗粒的MoO3/ZnMoO4棒状结构,棒的直径为0.42-0.47 μm,长径比为17-17.5:1,颗粒粒径为0.18-0.20 μm。
实施例4
(1)将6 mL的HNO3、24 mL的H2O和0.18 mL异丙醇混合均匀,然后加入0.5066 g的H24Mo7N6O24
(2)将得到混合溶液转移至反应釜中,密封后放入真空干燥箱,设置参数为:200℃,保温21.5 h,然后离心、洗涤、干燥得到MoO3;
(3)将得到的0.2500 g的MoO3加入到17 mL甲醇和0.13 mL丙三醇的溶液中,经过超声后,加入0.1700 g的PVP、0.0013 g的1-乙烯基-2-吡咯烷酮和0.0744 g的Zn (NO3)2
(4)将0.0821 g的2-甲基咪唑、0.0085 g的苯并咪唑溶解到17 mL甲醇和0.13 mL丙三醇的混合溶液中,搅拌均匀,得到B溶液;
(5)将B溶液加入到A溶液中,搅拌4.5 h后,经过洗涤、干燥后,得到前驱体;
(6)将前驱体置于马弗炉中,在空气气氛下按照3 ℃/min的升温速度由室温升至500 ℃,保温5 h,样品冷却后得到表面负载颗粒的MoO3/ZnMoO4棒状结构,棒的直径为0.50-0.58 μm,长径比为16-17:1,颗粒粒径为0.20-0.23 μm。
实施例5
(1)将3.5 mL的HNO3、19 mL的H2O和0.14 mL异丙醇混合均匀,然后加入0.4325 g的H24Mo7N6O24
(2)将得到混合溶液转移至反应釜中,密封后放入真空干燥箱,设置参数为:160℃,保温12 h,然后离心、洗涤、干燥;
(3)将得到的0.2500 g的MoO3加入到18 mL甲醇和0.15 mL丙三醇的溶液中,经过超声后,加入0.1200 g的PVP、0.0015 g的1-乙烯基-2-吡咯烷酮和0.0892 g的Zn (NO3)2
(4)将0.1478 g的2-甲基咪唑、0.0055 g的苯并咪唑溶解到18 mL甲醇和0.15 mL丙三醇的混合溶液中,搅拌均匀,得到B溶液;
(5)将B溶液加入到A溶液中,搅拌1 h后,经过洗涤、干燥后,得到前驱体;
(6)将前驱体置于马弗炉中,在空气气氛下按照1 ℃/min的升温速度由室温升至550 ℃,保温8 h,样品冷却后得到表面负载颗粒的MoO3/ZnMoO4棒状结构,棒的直径为0.60-0.65 μm,长径比为16.5-16.7:1,颗粒粒径为0.22-0.25 μm。
实施例6
(1)将4.5 mL的HNO3、22 mL的H2O和0.12mL异丙醇混合均匀,然后加入0.4572 g的H24Mo7N6O24
(2)将得到混合溶液转移至反应釜中,密封后放入真空干燥箱,设置参数为:170℃,保温18 h,然后离心、洗涤、干燥得到MoO3;
(3)将得到的0.25000 g的MoO3加入到13 mL甲醇和0.14 mL丙三醇的溶液中,经过超声后,加入0.1300 g的PVP、0.0017 g的1-乙烯基-2-吡咯烷酮和0.1041 g的Zn (NO3)2
(4)将0.1971 g的2-甲基咪唑、0.0136 g的苯并咪唑溶解到13 mL甲醇和0.14 mL丙三醇的混合溶液中,搅拌均匀,得到B溶液;
(5)将B溶液加入到A溶液中,搅拌2 h后,经过洗涤、干燥后,得到前驱体;
(6)将前驱体置于马弗炉中,在空气气氛下按照1 ℃/min的升温速度由室温升至570 ℃,保温6 h,样品冷却后得到表面负载颗粒的MoO3/ZnMoO4棒状结构,棒的直径为0.65-0.70 μm,长径比为14.5-15:1,颗粒粒径为0.25-0.29 μm。
对比例1
(1)将4 mL的HNO3、20 mL的H2O和0.15 mL异丙醇混合均匀,然后加入0.5000 g的氯化钼和0.0010 g的三甲基十八烷基氯化铵搅拌均匀,得到混合溶液;
(2)同实施例1步骤(2);
(3)同实施例1步骤(3);
(4)同实施例1步骤(4);
(5)同实施例1步骤(5);
(6)同实施例1步骤(6);
所得产物随炉冷却后得到了分散性差的片状结构,片的边长为0.21-0.40 μm,厚度为20-40 nm,不再具有表面负载颗粒的MoO3/ZnMoO4棒状结构。由此可以看出,钼源的种类对产物形貌具有重要影响。
对比例2
(1)将4 mL的HNO3、20 mL的H2O和0.15 mL异丙醇混合均匀,然后加入0.5000 g的H24Mo7N6O24
(2)同实施例1步骤(2);
(3)将得到的0.25 g的MoO3溶解到15 mL的水中,经过超声后,加入0.0149 g的Zn(NO3)2
(4)将0.0164 g的2-甲基咪唑、0.0010 g的苯并咪唑溶解到15 mL的水中,搅拌均匀,得到B溶液;
(5)同实施例1步骤(5);
(6)同实施例1步骤(6);
所得产物为表面光滑、分散性差的MoO3棒状结构,不再具有表面负载颗粒的MoO3/ZnMoO4棒状结构。由此可以看出,PVP和1-乙烯基-2-吡咯烷酮的引入对产物的物相组成和微观形貌具有重要影响。
对比例3
(1)同实施例1步骤(1);
(2)同实施例1步骤(2);
(3)同实施例1步骤(3);
(4)将步骤(3)得到的溶液A干燥后置于马弗炉中,在空气气氛下按照1 ℃/min的升温速度由室温升至450 ℃,保温2 h,样品随炉冷却后得到产物;
所得产物为表面光滑、分散性差的MoO3棒状结构,不再具有表面负载颗粒的MoO3/ZnMoO4棒状结构。由此可以看出,2-甲基咪唑和苯并咪唑的引入对产物的物相组成和微观形貌具有重要影响。
对比例4
(1)同实施例1步骤(1);
(2)同实施例1步骤(2);
(3)同实施例1步骤(3);
(4)同实施例1步骤(4);
(5)同实施例1步骤(5);
(6)将前驱体置于马弗炉中,在空气气氛下按照1 ℃/min的升温速度由室温升至400 ℃,保温0.5 h,样品随炉冷却后得到产物;
所得产物为有机无机成分复合的核壳棒状结构,不再具有表面负载颗粒的MoO3/ZnMoO4棒状结构。由此可以看出,热处理的温度与保温时间对产物的物相组成和微观形貌具有重要作用。
对比例5
(1)将1.0 mL的HNO3、22.15 mL的H2O和1.0 mL异丙醇混合均匀,然后加入0.5000 g的H24Mo7N6O24
(2)同实施例1步骤(2);
(3)同实施例1步骤(3);
(4)同实施例1步骤(4);
(5)同实施例1步骤(5);
(6)同实施例1步骤(6);
所得产物为类球形MoO3/ZnMoO4颗粒,颗粒尺寸为85-320 nm,不再具有表面负载颗粒的MoO3/ZnMoO4棒状结构。由此可以看出,HNO3、H2O和异丙醇的加入比例对产物的微观形貌具有重要作用。
一种表面负载颗粒的棒状MoO/ZnMoO结构的合成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0