

IPC分类号 : C07B53/00,C07B41/02,C07C29/145,C07C35/21,C07C35/50,C07C35/52,C07C41/26,C07C43/23








专利摘要
本发明涉及一种手性2‑芳亚甲基环烷醇及其不对称合成方法,具体结构如II所示。该方法以单磺酰手性二胺与金属钌、铑、铱的配合物为催化剂,甲酸/三乙胺的共沸物为氢源,在温和条件下通过对2‑芳亚甲基环烷酮(I)进行化学选择性不对称转移氢化制备手性2‑芳亚甲基环烷醇(II)。该方法操作简便,原料易得、收率和对映选择性都较高。
权利要求
1.一种手性2-芳亚甲基环烷醇的不对称合成方法,其特征在于,手性2-芳亚甲基环烷醇的结构式为
其中,Ar选自
R是氢、C1-C10烷基、C1-C10烷基氧基、卤代烷基、卤素、羟基、氨基、硝基、氰基;手性二胺金属络合物为催化剂,甲酸/三乙胺共沸物为氢源,在25-35℃下2-芳亚甲基环烷酮(I)通过不对称转移氢化得到手性2-芳亚甲基环烷醇(II);
上面给出的化合物I或II的定义中,所用术语不论单独使用还是用在复合词中,代表如下取代基:
卤素:指氟、氯、溴、碘;
烷基:指直链或支链烷基;
卤代烷基:指直链或支链烷基,在这些烷基上的氢原子部分或全部被卤原子取代,
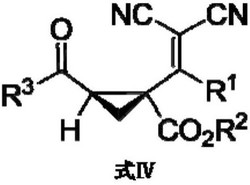
所述的催化剂为(R,R)-或(S,S)-N-单磺酰-二芳基手性乙二胺与过渡金属钌、铑或者铱的配合物,其结构通式如式III或式IV所示,
M为Ru、Rh或Ir;
所述结构通式III或IV中的Ar为苯基或对甲氧基、甲基取代的苯基;
所述结构通式III或IV中的R为-CH
R’为H、CH
L为苯、1,4-二甲基苯、1-甲基-4-异丙基苯、1,3,5-三甲基苯、1,2,3,4,5-五甲基苯、1,2,3,4,5,6-六甲基苯或五甲基环戊二烯;
X为Cl
Y为C、O。
2.根据权利要求1所述的手性2-芳亚甲基环烷醇的不对称合成方法,其特征在于,结构式为如下:
3.根据权利要求1所述的手性2-芳亚甲基环烷醇的不对称合成方法,其特征在于,所述的催化剂为下式中的任意一种:
4.根据权利要求3所述的手性2-芳亚甲基环烷醇的不对称合成方法,其特征在于,所述的催化剂为
5.根据权利要求1所述的手性2-芳亚甲基环烷醇的不对称合成方法,其特征在于,所述的氢源三乙胺/甲酸以任意比例的混合物。
6.根据权利要求1所述的手性2-芳亚甲基环烷醇的不对称合成方法,其特征在于,所述的溶剂为水、甲醇、乙醇、异丙醇、二氯甲烷、氯仿、1,2-二氯乙烷、苯、甲苯、二甲苯、四氢呋喃、二氧六环,二甲基亚砜、N,N-二甲基甲酰胺,以及一种或多种上述有机溶剂任意比例的混合物。
7.根据权利要求1所述的手性2-芳亚甲基环烷醇的不对称合成方法,其特征在于,催化剂:氢源:原料的摩尔比0.02~0.04:5~10:1。
说明书
技术领域
本发明属于不对称催化技术领域,具体涉及一种手性2-芳亚甲基环烷醇及其不对称合成方法。
背景技术
2-芳基亚甲基环烷醇是合成许多天然产物、药物、农药、生物活性物质的中间体。比如非甾体抗炎药洛索洛芬在用于慢性风湿性关节炎、变形性关节病、腰痛、肩关节周围炎、颈肩腕综合征以及手术后、外伤后、拔牙后的镇痛和消炎等方面有着广泛的应用,(S)-2-(4-溴苯基)亚甲基环戊醇是洛索洛芬的活性成分的关键手性中间体(Org.Lett.,2009,11(5),1103–1106)。
开发操作简单、经济、反应条件温和的手性2-芳基亚甲基环烷醇的不对称合成新方法具有重要研究意义和应用价值。目前文献报道了一种不对称氢化的方法,采用手性膦氮配体与铱的络合物为催化剂,以氢气为氢源通过不对称氢化来制备(J.Am.Chem.Soc.,2010,132(13),4538–4539)。该方法所用手性配体价格昂贵,对空气敏感,需要高压氢化设备和易燃的氢气,因此,开发温和条件下更实用的合成方法具有重要的需求。
不对称转移氢化具有操作简单,不需要氢气和高压设备等优势,在手性醇的不对称合成中越来越受到重视。手性二胺配体比手性膦氮配体稳定、合成简单、价格相对便宜,在不对称转移氢化反应中深受欢迎。
发明内容
本发明涉及一种手性2-芳亚甲基环烷醇及其不对称合成方法,所述的合成方法是以单磺酰手性二胺与金属钌、铑、铱的配合物为催化剂,甲酸/三乙胺的共沸物为氢源,在温和的条件下2-芳亚甲基环烷酮(I)通过不对称转移氢化得到手性2-芳亚甲基环烷醇(II),其结构式为
其中,Ar选自
R是氢、C1-C10烷基、C1-C10烷基氧基、卤代烷基、卤素、羟基、氨基、硝基、氰基。
上述结构式中,优选的结构式包括如下: 中的任意一种。
该结构式的合成工艺如下:
其中,Ar选自
R是氢、C1-C10烷基、C1-C10烷基氧基、卤代烷基、卤素、羟基、氨基、硝基、氰基;
上面给出的化合物I或II的定义中,所用术语不论单独使用还是用在复合词中,代表如下取代基:
卤素:指氟、氯、溴、碘;
烷基:指直链或支链烷基;
卤代烷基:指直链或支链烷基,在这些烷基上的氢原子部分或全部被卤原子取代。
本发明提供的不对称合成方法中所涉及的手性催化剂为(R,R)-或(S,S)-N-单磺酰-二芳基手性乙二胺与过渡金属钌、铑或者铱的配合物,其结构如下A-K,进一步优选为:催化剂E。
本发明提供的不对称合成方法中所涉及的氢源为不同比例的甲酸/三乙胺混合物,或甲酸钠,进一步优选为:甲酸/三乙胺=1.1:1;
本发明提供的不对称合成方法中所涉及的溶剂为水、甲醇、乙醇、异丙醇、二氯甲烷、氯仿、1,2-二氯乙烷、苯、甲苯、二甲苯、四氢呋喃、二氧六环,二甲基亚砜、N,N-二甲基甲酰胺,以及一种或多种上述有机溶剂任意比例的混合物,进一步优选为:三氯甲烷;
本发明提供的不对称合成方法反应温度优选为30℃;反应时间优选为6h;
本发明首次将不对称转移氢化应用到2-芳亚甲基环烷醇的不对称合成,与现有技术相比具有如下优点:所用手性二胺配体对空气和水稳定、合成简单、价格相对便宜、市场上可以买到;实验操作方便、安全、反应条件温和等。利用本发明提供的方法所得到的手性产物,是重要的医药或手性配体的中间体,因此本发明具有重要的工业应用价值。
具体实施方式
下面结合具体实施例,对本发明作进一步说明,但本发明并不限于以下实施例。
实施例1:(E)-2-苯基亚甲基环己醇的合成
将0.2mmol(E)-2-苯基亚甲基环己酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺,加入0.004mmol催化剂,1mL溶剂,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,柱层析分离得产物(石油醚:乙酸乙酯=10:1),HPLC测定对映体过量(ee)值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=90:10(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=6.41分钟,t2=7.79分钟;
实施例2:(E)-2-(3-甲氧基亚苄基)环己醇的合成
将0.2mmol(E)-2-(3-甲氧基亚苄基)环己酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率86%(石油醚:乙酸乙酯=10:1),HPLC测定对映体过量82%的ee值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=90:10(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=10.12分钟,t2=14.05分钟;
实施例3:(E)-2-(2-甲基亚苄基)环己醇的合成
将0.2mmol(E)-2-(2-甲基亚苄基)环己酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率80%(石油醚:乙酸乙酯=10:1),HPLC测定对映体过量83%的ee值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=90:10(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=5.87分钟,t2=6.86分钟;
实施例4:(E)-2-(4-叔丁基亚苄基)环己醇的合成
将0.2mmol(E)-2-(4-叔丁基亚苄基)环己酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率90%(石油醚:乙酸乙酯=8:1),HPLC测定对映体过量93%的ee值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=99:1(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=27.89分钟,t2=30.35分钟;
实施例5:(E)-2-(2-氯亚苄基)环己醇的合成
将0.2mmol(E)-2-(2-氯亚苄基)环己酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率72%(石油醚:乙酸乙酯=8:1),HPLC测定对映体过量76%的ee值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=90:10(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=5.89分钟,t2=8.02分钟;
实施例6:(E)-2-苯基亚甲基环庚醇的合成
将0.2mmol(E)-2-苯基亚甲基环庚酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率86%(石油醚:乙酸乙酯=8:1),HPLC测定对映体过量85%的ee值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=90:10(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=6.01分钟,t2=7.20分钟;
实施例7:(E)-2-(2-氟亚苄基)环庚醇的合成
将0.2mmol(E)-2-(2-氟亚苄基)环庚酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率86%(石油醚:乙酸乙酯=8:1),HPLC测定对映体过量90%的ee值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=90:10(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=5.06分钟,t2=5.59分钟;
实施例8:(E)-2-苯基亚甲基环戊醇的合成
将0.2mmol(E)-2-苯基亚甲基环戊酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率86%(石油醚:乙酸乙酯=8:1),HPLC测定对映体过量99%的ee值。HPLC分离条件:手性柱OJ-H柱,流动相:正己烷/异丙醇=98:2(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=36.81分钟,t2=40.40分钟;
实施例9:(E)-2-(2-氟亚苄基)环戊醇的合成
将0.2mmol(E)-2-(2-氟亚苄基)环戊酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率76%(石油醚:乙酸乙酯=8:1),HPLC测定对映体过量98%的ee值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=90:10(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=6.19分钟,t2=6.97分钟;
实施例10:(E)-2-(2-萘亚苄基)环己醇的合成
将0.2mmol(E)-2-(2-萘亚苄基)环己酮加到10毫升的密封试管中,加入1mmol甲酸/三乙胺(1.1:1),加入0.004mmol催化剂E,氯仿1mL,密封试管,30℃反应6小时。反应结束后用水洗,水相用乙酸乙酯萃取3次,合并有机相浓缩至干,分离产率85%(石油醚:乙酸乙酯=8:1),HPLC测定对映体过量83%的ee值。HPLC分离条件:手性柱OD-H柱,流动相:正己烷/异丙醇=90:10(体积比),流速:1.0毫升/分钟,波长:254纳米,柱温:30摄氏度,t1=8.37分钟,t2=9.80分钟;
一种手性2-芳亚甲基环烷醇及其不对称合成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。







![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)





动态评分
0.0