








专利摘要
本发明公开了两个基于双席夫碱配体的单核镝配合物及其制备方法和应用。这两个配合物分别为配合物1或配合物2,其中配合物1的化学式为[Dy(H3L)2Cl2]·EtOH·Cl,配合物2的化学式为[Dy(H3L)2Cl2H2O]·3H2O·Cl,其中H3L表示1,3‑二(2‑羟基萘亚甲基胺基)‑丙烷‑2‑醇;配合物属单斜晶系,I2/c空间群;配合物2属正交晶系,P212121空间群。本发明所述两个配合物制备方法简单、产率较高、重复性好,且均具有场诱导的单离子磁体行为,可以用于制备磁性材料。
权利要求
1.基于双席夫碱配体的单核镝配合物,为配合物1或配合物2,其中:
配合物1的化学式为:[Dy(H
该配合物属于单斜晶系,I2/c空间群,晶胞参数为:
配合物2的化学式为:[Dy(H
该配合物属于正交晶系,P2
2.权利要求1所述的基于双席夫碱配体的单核镝配合物的制备方法,其特征在于:
配合物1的制备方法为:将2-羟基-1-萘甲醛、1,3-二氨基-2-丙醇和DyCl
配合物2的制备方法为:将2-羟基-1-萘甲醛、1,3-二氨基-2-丙醇、DyCl
3.根据权利要求2所述的制备方法,其特征在于:反应在≥50℃的条件下进行。
4.根据权利要求2所述的制备方法,其特征在于:反应在60-90℃的条件下进行。
5.权利要求1所述的基于双席夫碱配体的单核镝配合物在制备磁性材料中的应用。
说明书
技术领域
本发明涉及基于双席夫碱配体的单核镝配合物及其制备方法和应用,属于磁性材料技术领域
背景技术
配位分子簇因配位构型的多变性和电子结构的复杂性决定了其在光、电、磁、催化等方面的潜在应用价值,越来越多的化学研究人员致力于对配位分子簇新颖结构的设计和丰富性能的追求,推动了配位化学与其他学科的交叉融合。
有关稀土Dy(III)分子磁性材料的研究一直以来都饱受科学研究者们的青睐,因为此类材料在电子设备、电子线路以及高密度存储方面具有潜在的应用价值。特别是因为它们有趣的磁学性质,稀土基单分子磁体作为磁性材料的一种,扮演着重要角色,在其领域内收到广泛关注,吸引了诸多科学研究者们研究的兴趣。同时,磁性材料的应用也非常的广泛,例如:能源、电信、自动控制、通讯、生物、医疗卫生等。随着信息化时代的发展,对磁性材料的要求也随之增加,要求磁性材料制造的器件不仅要大容量、小型化、高速度,而且还有要具有可靠性、耐久性和低成本的特点。此外,磁性材料以应用磁学技术为理论基础,与其他科学技术相互渗透、交叉,已逐步成为现代高新技术群体中不可缺少的部分。特别是纳米磁性材料在信息技术领域中日益显示出来的巨大经济效益和社会效益。因此,通过不同的有机配体构建具有SIM(单离子磁体)行为的新型磁性可调Dy(III)配合物具有重要意义。由于不同有机配体的配位能力及其得失电子与金属离子能级相比,开发具有SIM行为的可调发射型发光Dy(III)复合物仍然是一项具有挑战性的任务。
发明内容
本发明要解决的技术问题是提供两个具有单离子磁体行为的配位数可调的基于双席夫碱配体的单核镝配合物及其制备方法和应用。
本发明所述的基于双席夫碱配体的单核镝配合物,为配合物1或配合物2,其中:
配合物1的化学式为:[Dy(H3L)2Cl2]·EtOH·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇;
该配合物属于单斜晶系,I2/c空间群,晶胞参数为: α=90.00o,β=106.251(4)o,γ=90.00o;
配合物2的化学式为:[Dy(H3L)2Cl2H2O]·3H2O·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇;
该配合物属于正交晶系,P212121空间群,晶胞参数为: α=90.00o,β=90.00o,γ=90.00o。
本发明还提供上述基于双席夫碱配体的单核镝配合物的制备方法,具体的:
配合物1的制备方法为:将2-羟基-1-萘甲醛、1,3-二氨基-2-丙醇和DyCl3·6H2O置于第一混合溶剂中,溶解,所得溶液于加热条件下反应,即得;其中,所述的第一混合溶剂为乙腈与乙醇或甲醇的组合物;
配合物2的制备方法为:将2-羟基-1-萘甲醛、1,3-二氨基-2-丙醇、DyCl3·6H2O和Ni(OAC)2·4H2O置于第二混合溶剂中,溶解,所得溶液于加热条件下反应,即得;其中,所述的第二混合溶剂为乙腈与甲醇的组合物。
上述配合物1的制备方法中,2-羟基-1-萘甲醛、1,3-二氨基-2-丙醇和DyCl3·6H2O的摩尔比为化学计量比,在实际操作过程中,DyCl3·6H2O的量可相对过量一些。在第一混合溶剂的组成中,乙腈与乙醇,或者是乙腈与甲醇的体积比均优选为1:1-5,更优选为1:3-4。所述第一混合溶剂的用量可根据需要确定,通常以能溶解参加反应的原料为宜。具体地,以1mmol的2-羟基-1-萘甲醛为基准计算,全部原料所用混合溶剂的总用量通常为8-12mL。在具体溶解的步骤中,可将各原料分别用混合溶剂中的某一种组分溶解,然后再混合在一起反应;也可将所有原料混合在一起后再加混合溶剂溶解。
上述配合物2的制备方法中,2-羟基-1-萘甲醛、1,3-二氨基-2-丙醇和DyCl3·6H2O的摩尔比为化学计量比,在实际操作过程中,DyCl3·6H2O的量可相对过量一些。所述Ni(OAC)2·4H2O起到催化剂的作用,在不加Ni(OAC)2·4H2O时,反应没有配合物2生成。在第二混合溶剂的组成中,乙腈与甲醇的体积比优选为1:1-5,更优选为1:2-4。所述第二混合溶剂的用量可根据需要确定,通常以能溶解参加反应的原料为宜。具体地,以1mmol的2-羟基-1-萘甲醛为基准计算,全部原料所用混合溶剂的总用量通常为8-12mL。在具体溶解的步骤中,可将各原料分别用混合溶剂中的某一种组分溶解,然后再混合在一起反应;也可将所有原料混合在一起后再加混合溶剂溶解。
在本发明所述的配合物1和配合物2的制备方法中,反应优选是在≥50℃的条件下进行,在上述温度条件下进行反应的时间通常控制在72-100h。反应更优选是在60-90℃条件下进行。
本发明所述的配合物1和配合物2的制备方法中,在上述优选的溶剂组成以及反应温度条件下,可以获得更高的产率,且所得晶体的质量更好。
申请人对本发明所述的基于双席夫碱配体的单核镝配合物的磁性研究发现,该配合物的磁学性质表现为典型的单离子磁体行为。因此,本发明还包括上述单核镝配合物在制备磁性材料中的应用。
与现有技术相比,本发明提供了两个结构新颖的基于双席夫碱配体的单核镝配合物及其制备方法,申请人还发现它们为具有单分子磁体行为的可调发射型发光镝配合物,可以用于制备磁性材料;此外,该镝合物的制备方法简单、成本低廉、重复性好。
附图说明
图1为本发明实施例1制得的最终产物的化学结构图;
图2为本发明实施例1制得的最终产物的粉末衍射图;
图3为本发明实施例5制得的最终产物的化学结构图;
图4为本发明实施例5制得的最终产物的粉末衍射图;
图5为本发明实施例1制得的最终产物的红外光谱图;
图6为本发明实施例5制得的最终产物的红外光谱图;
图7为本发明实施例1制得的最终产物的热重曲线图;
图8为本发明实施例5制得的最终产物的热重曲线图;
图9为配合物1和2的χMT-T直流磁化率曲线图,其中(a)为配合物1的χMT-T直流磁化率曲线图,(b)为配合物2的χMT-T直流磁化率曲线图;
图10为配合物1和2的M-HT-1直流磁化率曲线图,其中(a)为配合物1的M-HT-1直流磁化率曲线图,(b)为配合物2的M-HT-1直流磁化率曲线图;
图11为配合物1和2在1000Oe直流场中不同温度的实部(χ')和虚部(χ”)变频交流磁化率依赖性曲线,其中(a)为配合物1在1200Oe直流场中不同温度的实部(χ')和虚部(χ”)变频交流磁化率依赖性曲线,(b)为配合物2在1000Oe直流场中不同温度的实部(χ')和虚部(χ”)变频交流磁化率依赖性曲线;
图12为配合物1和2从交流磁化率中获得的温度依赖性弛豫时间生成的阿伦尼乌斯方程图Hdc=1200和1000Oe,其中(a)为配合物1从交流磁化率中获得的温度依赖性弛豫时间生成的阿伦尼乌斯方程图,(b)为配合物2从交流磁化率中获得的温度依赖性弛豫时间生成的阿伦尼乌斯方程图。
具体实施方式
下面结合具体实施例对本发明作进一步的详述,以更好地理解本发明的内容,但本发明并不限于以下实施例。
实施例1:配合物1的制备
取0.1mmol的2-羟基-1-萘甲醛(0.0172g)用0.5mL乙腈溶解,得到溶液A;取0.05mmol的1,3-二氨基-2-丙醇(0.0045g)用1.5mL乙醇溶解(乙腈和乙醇的体积比为1:3),得到溶液B;将溶液A和溶液B加到含有0.1mmol DyCl3·6H2O(0.0368g)一端封闭的Pyrex管中,将Pyrex管抽真空,并将其另一端封口;将封好的Pyrex管置于80℃条件下反应72h,取出,缓慢冷却至室温,可观察到Pyrex管底部有黄色块状晶体析出。产率为23%(基于Dy)。
对本实施例所得产物进行表征:
1)单晶衍射及结构解析:
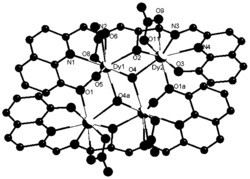
选取尺寸适中的黄色块状晶体置于安捷伦公司SuperNova单晶衍射仪上,采用石墨单色化的Mo-Kα 射线进行单晶测试。本实施例所得产物的初始晶体结构均采用SHELXS-97直接法解出,几何加氢,非氢原子坐标及各向异性热参数采用SHELXL-97均经全矩阵最小二乘法精修。所得晶体学和结构精修数据如下述表1所示,部分键长键角数据如下述表2所示,所得浅黄色块状晶体的化学结构如图1所示。
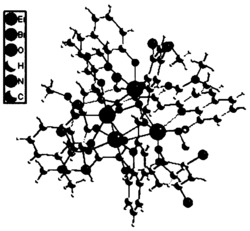
如图1所示,本实施例所得产物的空间群是I2/c,不对称单元由一个Dy,两个席夫碱配体,两个氯原子组成。金属中心(Dy)与螯合配体提供的四个氧原子和两个氯原子发生配位。
表1:配合物1和2的晶体学数据
表2:配合物1的部分部分键长/ 和键角/°数据
2)粉末衍射分析
为了研究所得产物的大量样品与单颗晶体的统一性,即纯相物质。本申请人将所得产物在常温条件下利用粉末衍射仪进行测试,测试范围为5-50°,扫描速率为5°/min。然后通过mercury软件模拟,将所得产物单晶结构的CIF文件模拟得出粉末图谱,再与实际谱图相比较,可以看出特征峰的位置及峰型基本一致,表明大量物质为纯相。所得产物的粉末衍射谱图如图2所示。
通过上述表征,确定所得的黄色块状晶体为本发明所述的配合物1即[Dy(H3L)2Cl2]·EtOH·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇。
对比例1-1
重复实施例1,不同的是,乙醇的用量改为0.25mL(乙腈和乙醇的体积比为1:0.5)。结果没有晶体或其它形状(如粉末)产物生成。
对比例1-2
重复实施例1,不同的是,乙醇的用量改为3.0mL(乙腈和乙醇的体积比为1:6)。结果没有晶体或其它形状(如粉末)产物生成。
实施例2:配合物1的制备
重复实施例1,不同的是:用甲醇替代乙醇,且甲醇的用量改为1.0mL(乙腈和甲醇的体积比为1:2),反应在50℃条件下进行,其它不变。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为9%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物1即[Dy(H3L)2Cl2]·EtOH·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇。
实施例3:配合物1的制备
重复实施例1,不同的是:乙醇的用量改为0.5mL(乙腈和乙醇的体积比为1:1),反应在90℃条件下进行,其它不变。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为14%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物1即[Dy(H3L)2Cl2]·EtOH·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇。
实施例4:配合物1的制备
重复实施例1,不同的是:乙醇的用量改为2.5mL(乙腈和乙醇的体积比为1:5),其它不变。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为15%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物1即[Dy(H3L)2Cl2]·EtOH·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇。
实施例5:配合物2的制备
取0.1mmol的2-羟基-1-萘甲醛(0.0172g)和0.05mmol的1,3-二氨基-2-丙醇(0.0091g)加到一端封闭的Pyrex管中,然后加入乙腈和甲醇的混合物(其中乙腈为0.5mL,乙腈和甲醇的体积比为1:3),溶解后,再加入0.1mmol的DyCl3·6H2O(0.0368g)和0.1mmol的Ni(OAC)2·4H2O(0.0199g),溶解后将Pyrex管抽真空,并将其另一端封口;将封好的Pyrex管置于80℃条件下反应72h,取出,缓慢冷却至室温,可观察到Pyrex管底部有淡黄色块状晶体析出。产率为26%(基于Dy)。
对本实施例所得产物进行表征:
1)单晶衍射及结构解析:
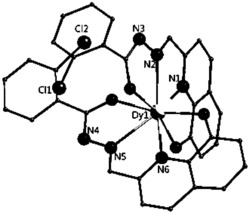
选取尺寸适中的淡黄色块状晶体置于安捷伦公司SuperNova单晶衍射仪上,采用石墨单色化的Mo-Kα 射线进行单晶测试。本实施例所得产物的初始晶体结构均采用SHELXS-97直接法解出,几何加氢,非氢原子坐标及各向异性热参数采用SHELXL-97均经全矩阵最小二乘法精修。所得晶体学和结构精修数据如上述表1所示,部分键长键角数据如下述表3所示,所得浅黄色块状晶体的化学结构如图4所示。
如图4所示,本实施例所得产物的空间群是P212121,不对称单元由一个Dy,两个席夫碱配体,两个氯原子和一个水分子组成。金属中心(Dy)与螯合配体提供的四个氧原子和两个氯原子以及一个水分子发生配位。
表3:配合物2的部分键长/ 和键角/°数据
2)粉末衍射分析
为了研究所得产物的大量样品与单颗晶体的统一性,即纯相物质。本申请人将所得产物在常温条件下利用粉末衍射仪进行测试,测试范围为5-50°,扫描速率为5°/min。然后通过mercury软件模拟,将所得产物单晶结构的CIF文件模拟得出粉末图谱,再与实际谱图相比较,可以看出特征峰的位置及峰型基本一致,表明大量物质为纯相。所得产物的粉末衍射谱图如图4所示。
通过上述表征,确定所得的淡黄色块状晶体为本发明所述的配合物2即[Dy(H3L)2Cl2(H2O)3]·3H2O·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇。
对比例2-1
重复实施例1,不同的是,用乙醇代替甲醇,其它不变。结果没有晶体或其它形状(如粉末)产物生成。
对比例2-2
重复实施例1,不同的是,甲醇的用量改为0.25mL(乙腈和乙醇的体积比为1:0.5)。结果没有晶体或其它形状(如粉末)产物生成。
对比例2-3
重复实施例1,不同的是,甲醇的用量改为3.0mL(乙腈和乙醇的体积比为1:6)。结果没有晶体或其它形状(如粉末)产物生成。
实施例6:配合物2的制备
重复实施例1,不同的是:甲醇的用量改为0.5mL(乙腈和甲醇的体积比为1:1),反应在50℃条件下进行,其它不变。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为12%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物2即[Dy(H3L)2Cl2H2O]·3H2O·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇。
实施例7:配合物2的制备
重复实施例1,不同的是:甲醇的用量改为2.5mL(乙腈和甲醇的体积比为1:5),反应在90℃条件下进行,其它不变。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为15%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物2即[Dy(H3L)2Cl2H2O]·3H2O·Cl,其中H3L表示1-({2-羟基-3-[(6-羟基-3-甲基-2-乙烯基-亚苄基)-氨基]-丙基亚胺基}-甲基)-萘-2-醇。
对本发明所述的配合物1和2(分别按实施例1和实施例5所述方法制得)进行荧光光谱分析:
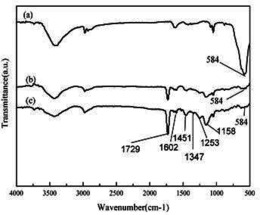
图5为配合物1的红外光谱图,在3431cm-1处有一个很宽的吸收峰,可以属为水分子ν(HO–H)的伸缩振动吸收峰;配合物在1636cm-1处的一个尖而且比较强的吸收峰,是配合物中配体上亚胺基团(–C=N–)的C=N伸缩振动吸收峰;配合物1548cm-1和1477cm-1的峰是芳环中C=N及C=C的伸缩振动吸收峰,也就是骨架谱带;在1150cm-1处的吸收峰,可以归属为配合物分子中配体内醇羟基C-O之间的伸缩振动吸收峰。
图6为配合物2的红外光谱图,在3432cm-1处有一个很宽的吸收峰,可以属为水分子ν(HO–H)的伸缩振动吸收峰;配合物在1635cm-1处的一个尖而且比较强的吸收峰,是配合物中配体上亚胺基团(–C=N–)的C=N伸缩振动吸收峰;配合物1549cm-1和1511cm-1的峰是芳环中C=N及C=C的伸缩振动吸收峰,也就是骨架谱带;在1133cm-1处的吸收峰,可以归属为配合物分子中配体内醇羟基C-O之间的伸缩振动吸收峰。
对本发明所述的配合物1和2(分别按实施例1和实施例5所述方法制得)进行热重分析:
2)热重分析
实验温度控制为室温到1000℃,流速为15cm3/min,升温速率为3℃/min,氮气条件下的热重分析表明:
配合物1在接近260℃时才开始分解,说明其热稳定性好。配合物1的热重分析图如图7所示。
配合物2在30-122℃开始发生第一个失重行为,失去总质量的4.8%,失去所有溶剂分子,这一失重过程基本对应于硝酸根的失去,测定值与计算值7.2%接近。从122℃到250℃金属骨架稳定存在,随后温度升高骨架开始坍塌,这一失重过程主要是有机配体的逐步热分解,最终剩下61.02%,残留物可能是金属的氧化物Dy2O3,计算值为58.43%。说明其热稳定性好。配合物2的热重分析图如图8所示。
对本发明所述的配合物1和2(分别按实施例1和实施例5所述方法制得)进行磁性分析:
在直流外磁场为1000Oe下,2–300K温度区间内,分别测量配合物1和2的摩尔磁化率随温度的变化的情况。配合物1和2的χM-T曲线类似,根据χm-T的曲线可以看出,300-180K磁化率几乎为零,180K以下,随温度的降低,磁化率逐渐增大。在外加直流场的作用下,配合物的摩尔磁化率在高温区几乎保持不变,在低温区迅速增大,这种现象符合一般分子磁体的顺磁行为,宏观上可以解释为,在高温区受热扰动的影响较大,磁距随机取向,随着温度的降低,热扰动的影响逐渐降低,磁距趋向于平行排列,磁化率逐渐增大。配合物1和2的χMT-T曲线类似,如图9所示,由图中黑色的χMT-T的曲线可知,在300K时,配合物1和2的实验测得的χMT的值分别为12.61cm3K mol-1和12.23cm3K mol-1,其实验值稍低于两个唯自旋的Dy(III)离子的理论值28.34cm3K mol-1:(一个自由的Dy(III)离子:14.17cm3Kmol-1,6H15/2,S=5/2,L=5,g=4/3),从300K到200K,χmT值随温度降低几乎保持不变,200-50K,χMT开始随温度降低,50-2K,降低极为明显,在2K时配合物1和2分别达到最小值9.43cm3Kmol-1和8.33cm3Kmol-1。
配合物1和2在外加场0-40kOe的条件下,各个温度的M-HT-1的曲线图如图10所示,实验数据表明,在低场下各个温度的M-HT-1曲线并没有重合,可归因于体系中存在强的磁各向异性和低激发态。对于配合物1随着外加磁场的增加,配合物的磁化强度迅速增大,最终各个温度的磁化强度趋于重合。配合物2不同于配合物1,最终各个温度的磁化强度没有达到饱和。例如在2K,40kOe下,1和2磁化强度分别为6.32μB和5.08μB这个数值比理论饱和值10μB(一个DyIII离子的磁化强度为10μB)要低,造成这种差异可能是由于DyIII离子在晶体场中诱导了斯塔克能级的分裂,消除了6H15/2基态的16重简并。
为了探索各向异性磁矩的磁化动力学,对两个配合物进行交流(AC)磁化率测试,配合物1在1200直流场和一个3Oe交流场中以1-1000Hz范围内的频率振荡测量,配合物2是在1000直流场和一个3Oe交流场中以1-1000Hz范围内的频率振荡测量。对于配合物1和2,χ”与F的关系曲线在不同频率下从4.2到1.8K测量;实部(χ')和虚部(χ”)部分都具有频率和温度依赖性,如图11所示。此外,在所有应用的频率中出现1和2的异相峰,表明预期会有更高的能垒。配合物1和2的χ”曲线的峰值以温度序列从低到高逐渐转移,说明两个化合物的χ”在选定的温度范围内总是表现出频率依赖性。在不同频率下测量的配合物1和2的同相(χ')和异相(χ”)交流磁化率彼此差异很大,揭示了SMM(单分子磁体)典型的慢磁弛豫的存在.在不同温度下测量的配合物1和2的频率依赖显示出彼此之间的差异很大。配合物1和2的χ”与v曲线表示在增加测量温度时移动到更高频率的最大值。相比之下,χ”与v曲线2的最大值出现在高于1的频率。在1000Oe直流场下,配合物1的最大值vs.χ”对比1曲线的频率低于1000Oe直流场的频率。所有这些特征都证实了配合物1和2都是SMM。
为了计算配合物1和2的能垒Ueff和驰豫时间τ0,两个化合物的AC-F磁测量数据用于绘制半圆形Cole-Cole曲线,如图11所示两种化合物的χ'和χ”磁化率在较低温度区域显示出显着的热依赖性最大值,显然,随着温度的升高,两种化合物中χ'曲线的最大值从较低频率缓慢移动到较高频率。此外,ln(τ)形式的磁化弛豫时间(τ)被描述为如图12中1/T的函数。根据高温曲线拟合Arrhenius定律的行为,可以获得有效屏障(Ueff/kB)。众所周知,基于镧系元素的SMM的弛豫机制可能涉及Orbach(τ0-exp(-Ueff/kBT)),拉曼(CTn),QTM(τQTM-1)和直接弛豫过程(AHmT)四个可能的过程。弛豫时间τ值取决于频率相关的交流磁化率,由阿伦尼乌斯拟合后的χ'最大值[τ=τ0exp(Ueff/kBT)]得出,配合物1和2的能垒和弛豫时间分别为Ueff=43.86K和τ0=3.65×10-6s和Ueff=21.57K和τ0=3.64×10-10s在1000和1200直流场下(如图12所示)。由此产生的能垒值和弛豫时间与已知的Dy(III)配合物不同。
基于双席夫碱配体的单核镝配合物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0