









专利摘要
本发明提供了一种皂苷分离纯化方法。采用二维制备液相色谱从皂苷提取物中高效分离纯化皂苷单体,第一维采用反相色谱柱,第二维采用亲水色谱柱。流动相组成为乙腈、甲醇、乙醇或水,无缓冲盐添加,便于样品制备后处理。采用线性梯度、台阶梯度或等度洗脱方式。选取三七叶水提物第一维制备所得三个有代表性的馏分,通过第二维亲水色谱制备获得八个皂苷单体,其中包括两对同分异构体和一个新皂苷。该方法可以实现皂苷的目标制备,可获取批量的已知活性皂苷,同时富集分离微量皂苷,从而不断丰富皂苷库,为皂苷类化合物的活性研究和单一成分的新药开发提供物质基础。
权利要求
1.一种皂苷分离纯化方法,其特征在于:采用二维制备液相色谱从皂苷提取物中分离纯化皂苷单体,第一维采用反相色谱柱,流动相组成为乙腈、甲醇或乙醇中的一种或两种与水混合,洗脱方式为线性梯度或台阶梯度;第二维采用亲水色谱柱,流动相组成为甲醇、乙醇或水中的一种或两种与乙腈混合,采用等度洗脱方式。
2.按照权利要求1所述皂苷分离纯化方法,其特征在于:所述反相色谱柱为C18 TDE、X4、XTerra MS C18或SunFire C18;所述亲水色谱柱为酰胺柱(XAmide)、两性离子柱(Click XIon)、麦芽糖柱(ClickMaltose)、环糊精柱(Clickβ-CD)、二醇基柱(Diol)或硅胶柱(Silica)。
3.按照权利要求1所述皂苷分离纯化方法,其特征在于:色谱操作参数如下:色谱柱内径为4.6-100mm;样品浓度为1mg/mL-1g/mL;进样量为1μL-40mL;流速为0.5-480mL/min;柱温为0-60℃。
4.按照权利要求1所述皂苷分离纯化方法,其特征在于:所述第一维制备流动相组成为乙腈-水、甲醇-水或乙醇-水;第二维制备流动相组成为乙腈-水、乙腈-甲醇、乙腈-乙醇、乙腈-甲醇-水或乙腈-乙醇-水。
5.按照权利要求1所述皂苷分离纯化方法,其特征在于:所述皂苷提取物为三七叶水提物。
6.一种权利要求5所述皂苷类化合物的制备方法,其特征在于:
1)三七叶水提物先经第一维制备型HPLC分离,色谱条件:色谱柱为C18 TDE柱;水(A)和乙腈(B)流动相体系;洗脱梯度为0-5min,体积浓度20%→32%B;5-45min,体积浓度32%→68%B;45-50min,体积浓度68%→100%B;50-55min,100%B;流速为300mL/min;检测波长为203nm;样品溶液浓度为300mg/mL,其采用体积浓度20%ACN-80%H2O溶解;进样体积为10mL;1-55min,每分钟收集一份,共计54个组分,每个组分浓缩至干后备用;
2)在第一维制备所得54个组分中,皂苷主要分布在Fractions 6-30共25个组分中;之后,针对这25个富含皂苷的组分进行第二维亲水色谱制备,色谱柱为酰胺柱;采用乙腈水流动相体系,根据各个组分的色谱保留特性,流动相中乙腈的体积浓度控制在60%-95%;采用等度洗脱方式制备皂苷单体。
7.一种权利要求6所述皂苷类化合物的制备方法,其特征在于:
选取第一维制备所得3个有代表性的馏分(Fr.9,Fr.11和Fr.13),通过第二维亲水色谱制备获得皂苷单体,化合物的具体制备过程如下:
选取Fraction 9采用亲水色谱模式进行第二维制备型HPLC分离,色谱条件:色谱柱为酰胺柱(XAmide);水(A)和乙腈(B)流动相体系;体积浓度76%B等度洗脱;流速为20mL/min;检测波长为203nm;进样量为1mL;收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P1和P2两个化合物;
选取Fraction 11采用亲水色谱模式进行第二维制备型HPLC分离,色谱条件:色谱柱为酰胺柱(XAmide);水(A)和乙腈(B)流动相体系;体积浓度80%B等度洗脱;流速为20mL/min;检测波长为203nm;进样量为1mL。收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P3、P4、P5和P6四个化合物;
选取Fraction 13采用亲水色谱模式进行第二维制备型HPLC分离,色谱条件:色谱柱为酰胺柱(XAmide);水(A)和乙腈(B)流动相体系;体积浓度80%B等度洗脱;流速为20mL/min;检测波长为203nm;进样量为1mL。收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P7和P8两个化合物;
第一维所得3个有代表性的馏分(Fr.9,Fr.11和Fr.13),通过第二维亲水色谱制备获得8个皂苷单体,包括2对同分异构体和1个新皂苷;
所述化合物的结构信息如下:
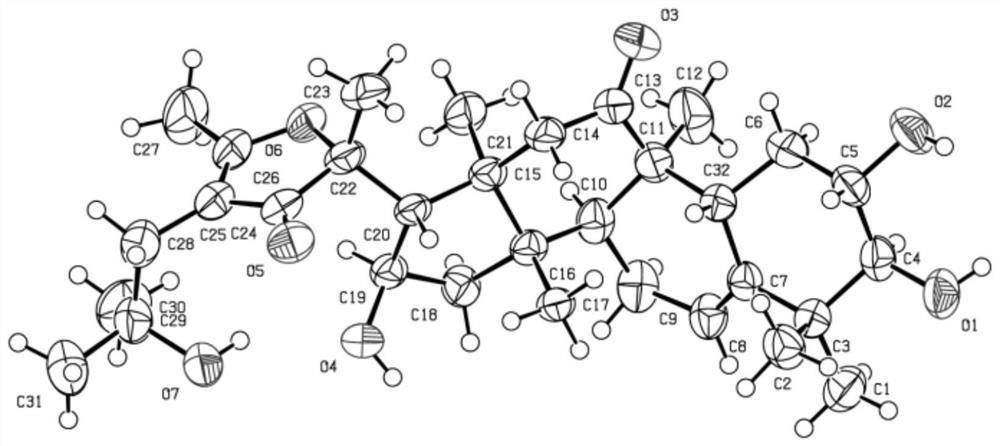
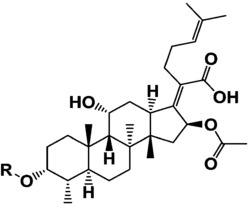
化合物P1为Notoginsenoside FP2,分子量为1210,分子式为C58H98O26,R1为O-glu(2→1)glu(2→1)xyl,R2为O-glu(6→1)araF;化合物P2为Notoginsenoside Fa,分子量为1240,分子式为C59H100O27,R1为O-glu(2→1)glu(2→1)xyl,R2为O-glu(6→1)glu;化合物P3为Ginsenoside Rc,分子量为1078,分子式为C53H90O22,R1为O-glu(2→1)glu,R2为O-glu(6→1)araF;化合物P4为Ginsenoside Rb1,分子量为1108,分子式为C54H92O23,R1为O-glu(2→1)glu,R2为O-glu(6→1)glu;化合物P5为Notoginsenoside Fc,分子量为1210,分子式为C58H98O26,R1为O-glu(2→1)glu(2→1)xyl,R2为O-glu(6→1)xyl;化合物P6为New Saponin,分子量为1342,分子式为C63H106O30,R1为O-glu(2→1)glu(2→1)xyl,R2为O-glu(6→1)araP(4→1)xyl;化合物P7为Ginsenoside Rd,分子量为946,分子式为C48H82O18,R1为O-glu(2→1)glu,R2为O-glu;化合物P8为Ginsenoside Rb3,分子量为1078,分子式为C53H90O22,R1为O-glu(2→1)glu,R2为O-glu(6→1)xyl。
说明书
技术领域
本发明涉及皂苷类化合物的分离纯化,具体地说是一种通过二维制备色谱分离技术从皂苷提取物中分离制备皂苷单体,包括常量已知皂苷、皂苷异构体和微量新皂苷等的方法。
背景技术
皂苷(Saponins)是苷元(Sapogenins)为三萜(Triterpenoids)或螺旋甾烷类(Spirostane)化合物的一类糖苷。皂苷根据苷元连接糖链数目的不同,可分为单糖链皂苷、双糖链皂苷及三糖链皂苷。在皂苷的化学结构中,由于苷元具有不同程度的亲脂性,糖链具有较强的亲水性,使皂苷成为一种表面活性剂,用力振摇其水溶液可产生持久性泡沫。
皂苷类化合物除具有表面活性、溶血、毒鱼等特性外,还具有多种生理功能,是许多中药的主要活性成分。皂苷的生物活性除与苷元有关外,与糖链的结构也关系密切。S.G.Sparg、王楠、张云峰和张存莉等分别对皂苷类化合物具有的生物活性进行了综述,这些活性主要包括防治心血管系统疾病、抗癌、降血糖、降血脂、保肝、抗炎、抗过敏、抗微生物、抗衰老、抗生育等(S.G.Sparg,M.E.Light,J.van Staden,Biological activities and distribution of plant saponins,Journal of Ethnopharmacology 94(2004)219-243;王楠.皂苷生物活性的研究进展,医学研究生学报,2007,20(2):211-214;张云峰,魏东,邓雁如,等.三萜皂苷的生物活性研究新进展,中成药,2006,28(9):1349-1353;张存莉,吴战库,马惠玲,等.甾体皂苷的生物活性研究进展,西北林学院学报,2003,18(2):95-100.)。
适应中药现代化的需要,为了揭示以皂苷类化合物为主要活性成分的中药治疗疾病的物质基础,科研人员开展了大量有关皂苷类化合物的分离纯化工作。采用的方法主要是使用甲醇或稀乙醇作提取溶剂,提取液回收溶剂后,用水稀释,经正丁醇萃取或大孔树脂纯化,得粗皂苷,最后用硅胶柱色谱进行分离或高效液相制备,得到单体,常用的洗脱剂有不同比例的氯仿-甲醇-水混合溶剂和水饱和的正丁醇。该方法主要存在以下问题:一是,皂苷类化合物极性较强,在正相洗脱液中的溶解性较差,导致其在硅胶柱分离时易产生拖尾和死吸附等问题;二是,皂苷类化合物一般只在紫外末端有吸收,这给制备高效液相色谱在线检测带来不便;三是,大量结构相似或同分异构体皂苷类化合物的聚类存在,对常规C18制备色谱的分离能力是一种挑战;四是,皂苷类化合物在水中浓缩时会产生大量泡沫,给反相色谱 柱后样品处理带来不便;五是,化合物制备可控性差,无法实现有效的目标制备,导致大量已知化合物的重复获得,造成人力、物力的极大浪费;六是,色谱分离模式僵化,分离选择性差,使得发现新化合物的困难越来越大。
五加科人参属中三种重要植物——人参(Panax ginseng C.A.Mey)、三七(Panax Notoginseng(Burk.)F.H.Chen)和西洋参(Panaxquinquefolium L.)中已发现的人参皂苷有100多种。主要包括达玛烷型的原人参二醇型(Protopanaxadiol)和原人参三醇型(Protopanaxatriol)、齐墩果烷型(Oleanane)以及奥克梯隆醇型(Ocotillol)皂苷。反相C18梯度洗脱是人参皂苷常用的分析方法,但分析时间一般较长。鉴于HILIC与RP分离机理完全不同,有研究报道采用HILIC分离模式等度洗脱分析人参皂苷类化合物,可有效缩短分析时间(Da Wei Lou,Yoshihiro Saito,Pawel K,Zarzycki,MitsuhiroOgawa,Kiyokatsu Jinno,Isocratic separation of ginsenosides byhigh-performance liquid chromatography on a diol column at subambienttemperatures,Anal.Bioanal.Chem.(2006)385:96-104.;N.S.Quiming,N.L.Denola,A.B.Soliev,Y.Saito,K.Jinno,High performance liquidchromatographic separation and quantitative analysis of ginsenosidesusing a polyvinyl alcohol-bonded stationary phase,Chromatographia2007,66,July(No.1/2).;Noel S.Quiming,Nerissa L.Denola,AzamjonB.Soliev,Yoshihiro Saito,Kiyokatsu Jinno,Retention behavior ofginsenosides on a poly(vinyl alcohol)-bonded stationary phase inhydrophilic interaction chromatography,Anal.Bioanal.Chem.(2007)389:1477-1488.)。因目前商品化的亲水色谱分离材料种类很有限,将其用于皂苷类化合物的制备还鲜有报道。此外,有研究报道采用高速逆流色谱(HSCCC)技术分离制备人参皂苷类化合物(Qizhen Du,Gerold Jerzc,Reiner Waibelb,Peter Winterhalterc,Isolation ofdammarane saponins from Panax notoginseng by high-speedcounter-current chromatography,J.Chromatogr.A 1008(2003)173-180.;Young Wan Ha,Soon Sung Lim,In Jin Ha,Yun-Cheol Na,Jung-Ju Seo,Heungsop Shin,Sung Ho Son,Yeong Shik Kim,Preparative isolation of four ginsenosides from Korean red ginseng(steam-treated Panax ginseng C.A.Meyer),by high-speedcounter-current chromatography coupled with evaporative light scatteringdetection,J.Chromatogr.A 1151(2007)37-44.;Yijun Cheng,QionglinLiang,Ping Hu,Yiming Wang,Frank Wu Jun,Guoan Luo,Combinationof normal-phase medium-pressure liquid chromatography andhigh-performance counter-current chromatography for preparation ofginsenoside-Ro from panax ginseng with high recovery and efficiency,Separation and Purification Technology 73(2010)397-402.),但该方法 主要适用于分离制备已知且高含量的化合物,而且适宜萃取溶剂体系的选择非常困难,萃取过程中易产生乳化现象给分离检测带来不便。
发明内容
本发明涉及一种皂苷的分离纯化方法。采用二维制备液相色谱从皂苷提取物中分离纯化皂苷单体,第一维采用反相色谱柱,第二维采用亲水色谱柱。流动相组成为乙腈、甲醇、乙醇或水,无缓冲盐添加,便于样品制备后处理。采用线性梯度、台阶梯度或等度洗脱方式。
其中所述反相色谱柱为C18 TDE、X4、XTerra MS C18或SunFire C18;所述亲水色谱柱为酰胺柱(XAmide)、两性离子柱(ClickXIon)、麦芽糖柱(Click Maltose)、环糊精柱(Clickβ-CD)、二醇基柱(Diol)或硅胶柱(Silica)。色谱操作参数如下:色谱柱内径为4.6-100mm;样品浓度为1mg/mL-1g/mL;进样量为1μL-40mL;流速为0.5-480mL/min;柱温为0-60℃。
所述第一维制备流动相组成为乙腈-水、甲醇-水或乙醇-水;第二维制备流动相组成为乙腈-水、乙腈-甲醇、乙腈-乙醇、乙腈-甲醇-水或乙腈-乙醇-水。
选取三七叶水提物第一维所得3个有代表性的馏分(Fr.9,Fr.11和Fr.13),通过第二维亲水色谱制备获得8个皂苷单体,包括2对同分异构体和1个新皂苷。所述化合物的结构信息如下:
glu:β-D-吡喃型葡萄糖基,xyl:β-D-吡喃型木糖基,araF:α-L-呋喃型阿拉伯糖基,araP:α-L-吡喃型阿拉伯糖基
所述化合物具体制备方法为:
三七叶水提物先经第一维制备型HPLC分离,色谱条件:色谱柱为C18 TDE柱(220×80mm,i.d.,10μm);水(A)和乙腈(B)流动相体系;洗脱梯度为0-5min,体积浓度20%→32%B;5-45min,体积浓度32%→68%B;45-50min,体积浓度68%→100%B;50-55min,100%B;流速为300mL/min;检测波长为203nm;样品溶液浓度为300mg/mL;进样体积为10mL。1-55min,每分钟收集一份,共计54个组分,每个组分浓缩至干后备用。
选取Fraction 9采用亲水色谱模式进行第二维制备型HPLC分离,色谱条件:色谱柱为酰胺柱(XAmide,250×20mm,i.d.,10μm);水(A)和乙腈(B)流动相体系;体积浓度76%B等度洗脱;流速为20mL/min;检测波长为203nm;进样量为1mL。收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P1和P2两个化合物。
选取Fraction 11采用亲水色谱模式进行第二维制备型HPLC分离,色谱条件:色谱柱为酰胺柱(XAmide,250×20mm,i.d.,10μm);水(A)和乙腈(B)流动相体系;体积浓度80%B等度洗脱;流速为20mL/min;检测波长为203nm;进样量为1mL。收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P3、P4、P5和P6四个化合物。
选取Fraction 13采用亲水色谱模式进行第二维制备型HPLC分离,色谱条件:色谱柱为酰胺柱(XAmide,250×20mm,i.d.,10μm);水(A)和乙腈(B)流动相体系;体积浓度80%B等度洗脱;流速为20mL/min;检测波长为203nm;进样量为1mL。收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P7和P8两个化合物。
该方法可以实现皂苷的目标制备,可获取批量的已知活性皂苷,同时富集分离微量皂苷,从而可以不断丰富皂苷库,为皂苷类化合物的活性研究和单一成分的新药开发提供物质基础。
具体实施方式
现结合实例,对本发明做进一步说明。实例仅限于说明本发明,而非对本发明的限定。
实施例1:皂苷提取物第一维馏分制备
三七叶水提物经制备型HPLC分离(C18 TDE柱;水(A)和乙腈(B)流动相体系;洗脱梯度为0-5min,体积浓度20%→32%B;5-45min,体积浓度32%→68%B;45-50min,体积浓度68%→100%B;50-55min,100%B);分离过程中采用紫外检测器检测,检测波长203nm。1-55min,每分钟收集一份,共计54个组分。分别为Fractions 1-54,皂苷主要集中在Fractions 6-30。
实施例2:化合物P1和P2的制备
选取Fraction 9采用亲水色谱模式进行第二维制备型HPLC分离(色谱柱为酰胺柱;水(A)和乙腈(B)流动相体系;体积浓度76%B等度洗脱)流速为20mL/min;检测波长为203nm;进样量为1mL。收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P1和P2两个化合物。HPLC检测纯度大于95%,经理化测定,数据如下:P1,白色粉末,HR-ESI-MS:[M+H]+(m/z):1211.6409.1H NMR(600MHz,pyridine-d5)δ:0.77(1H,s,H-19),0.92(2H,s,H-18,30),1.08(1H,s,H-29),1.25(1H,s,H-28),1.60(1H,s,H-21),1.62(1H,s,H-27),1.64(1H,s,H-26),3.28(1H,dd,J=4.2,11.4,H-3),5.29(1H,t,J=7.2,H-24),4.92(1H,d,J=7.8),5.50(1H,d,J=7.8),5.40(1H,d,J=6.6),5.13(1H,d,J=7.8),4.85(1H,brs).13C NMR:见表1.P2,白色粉末,HR-ESI-MS:[M+H]+(m/z):1241.6498.1H NMR(600MHz,pyridine-d5)δ:0.78(1H,s,H-19),0.93(1H,s,H-30),0.94(1H,s,H-18),1.08(1H,s,H-29),1.25(1H,s,H-28),1.58(1H,s,H-21),1.64(2H,s,H-26,27),3.28(1H,dd,J=4.2,11.4,H-3),5.29(1H,t,J=6.6,H-24),4.91(1H,d,J=7.8),5.49(1H,d,J=7.2),5.39(1H,d,J=7.2),5.12(1H,d,J=7.8),5.09(1H,d,J=7.8).13C NMR:见表1.
实施例3:化合物P3-P6的制备
选取Fraction 11采用亲水色谱模式进行第二维制备型HPLC分离(色谱柱为酰胺柱;水(A)和乙腈(B)流动相体系;体积浓度80%B等度洗脱)流速为20mL/min;检测波长为203nm;进样量为1mL。收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P3-P6四个化合物。HPLC检测纯度大于95%,经理化测定,数据如下:P3,白色粉末,HR-ESI-MS:[M+H]+(m/z):1079.5989.1H NMR(600MHz,pyridine-d5)δ:0.78(1H,s,H-19),0.92(1H,s,H-30),0.93(1H,s,H-18),1.08(1H,s,H-29),1.26(1H,s,H-28),1.60(1H,s,H-21),1.62(1H,s,H-27),1.64(1H,s,H-26),3.24(1H,dd,J=4.2,11.4,H-3),5.29(1H,t,J=6.6,H-24),4.90(1H,d,J=7.8),5.35(1H,d,J=7.8),5.12(1H,d,J=7.8),4.85(1H,brs).13C NMR:见表1.P4,白色粉末,HR-ESI-MS:[M+H]+(m/z):1109.6064.1H NMR(600MHz,pyridine-d5)δ:0.80(1H,s,H-19),0.94(2H,s,H-18,30),1.09(1H,s,H-29),1.27(1H,s,H-28),1.59(1H,s,H-21),1.64(2H,s,H-26,27),3.26(1H,dd,J=4.2, 11.4,H-3),5.30(1H,t,J=6.6,H-24),4.91(1H,d,J=7.8),5.36(1H,d,J=7.8),5.12(1H,d,J=7.8),5.09(1H,d,J=7.8).13C NMR:见表1.P5,白色粉末,HR-ESI-MS:[M+H]+(m/z):1211.6390.1H NMR(600MHz,pyridine-d5)δ:0.78(1H,s,H-19),0.92(1H,s,H-30),0.94(1H,s,H-18),1.09(1H,s,H-29),1.25(1H,s,H-28),1.59(1H,s,H-21),1.63(2H,s,H-26,27),3.28(1H,dd,J=4.2,12,H-3),5.29(1H,t,J=7.2,H-24),4.91(1H,d,J=7.8),5.50(1H,d,J=7.8),5.40(1H,d,J=7.2),5.12(1H,d,J=7.8),4.97(1H,d,J=7.2).13C NMR:见表1.P6,白色粉末,HR-ESI-MS:[M+H]+(m/z):1343.6930.1H NMR (600MHz,pyridine-d5)δ:0.78(1H,s,H-19),0.92(1H,s,H-30),0.95(1H,s,H-18),1.09(1H,s,H-29),1.25(1H,s,H-28),1.59(1H,s,H-21),1.62(1H,s,H-27),1.63(1H,s,H-26),3.28(1H,dd,J=4.2,11.4,H-3),5.28(1H,t,J=6.6,H-24),4.92(1H,d,J=6.0),5.50(1H,d,J=7.8),5.40(1H,d,J=6.6),5.10(1H,d,J=7.8),5.50(1H,d,J=7.8).13C NMR:见表1.
实施例4:化合物P7和P8的制备
选取Fraction 13采用亲水色谱模式进行第二维制备型HPLC分离(色谱柱为酰胺柱XAmide,250×20mm,i.d.,10μm);水(A)和乙腈(B)流动相体系;体积浓度80%B等度洗脱)流速为20mL/min;检测波长为203nm;进样量为1mL。收集各个色谱峰,分别回收溶剂,经核磁实验确定,获得P7和P8两个化合物。HPLC检测纯度大于95%,经理化测定,数据如下:P7,白色粉末,HR-ESI-MS:[M-H]-(m/z):945.5468.1H NMR(600MHz,pyridine-d5)δ:0.79(1H,s,H-19),0.93(1H,s,H-30),0.94(1H,s,H-18),1.09(1H,s,H-29),1.26(1H,s,H-28),1.57(2H,s,H-21,27),1.61(1H,s,H-26),3.25(1H,dd,J=4.2,12,H-3),5.22(1H,t,J=6.6,H-24),4.91(1H,d,J=7.8),5.35(1H,d,J=7.8),5.18(1H,d,J=7.2).13C NMR:见表1.P8,白色粉末,HR-ESI-MS:[M+H]+(m/z):1079.5948.1H NMR(600MHz,pyridine-d5)δ:0.78(1H,s,H-19),0.93(1H,s,H-30),0.95(1H,s,H-18),1.09(1H,s,H-29),1.26(1H,s,H-28),1.59(1H,s,H-21),1.63(2H,s,H-26,27),3.26(1H,dd,J=4.2,12,H-3),5.29(1H,t,J=6.6,H-24),4.90(1H,d,J=7.2),5.35(1H,d,J=7.8),5.11(1H,d,J=7.8),4.97(1H,d,J=7.8).13C NMR:见表1.
表1化合物P1-P8的13C NMR数据(150MHz,氘代吡啶)(The 13CNMR (150MHz)spectral data of the compounds P1-P8(in pyridine-d5))
glu:β-D-吡喃型葡萄糖基,xyl:β-D-吡喃型木糖基,ara:α-L-呋喃型阿拉伯糖基或α-L-吡喃型阿拉伯糖基(glu:β-D-glucopyranosyl,xyl:β-D-xylopyranosyl,ara:α-L-arabinofuranosyl or α-L-arabinopyranosyl)
一种皂苷分离纯化方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0