

IPC分类号 : C07J63/00,C07J75/00,A61K31/56,A61P1/16


![镉-有机超分子固体[Cd(ppa)(bpe)] n 及制备方法](https://zhichawang.com/youzhi/106279217A/106279217A.png)






专利摘要
本发明公开了一组三羟基取代五环三萜类化合物及其制备方法和应用,该化合物的通式为结构式I,其中:R1、R2为氢或甲基;R3选自-COOH、-CH2OH、CHO、-COOR1、-CONH2、-CONHR1、-CONR1R2中任一种;R1、R2选自含有1-8个碳原子的烷基、取代或未取代的苯基、取代或未取代的苯烷基中的一种,R2与R1可相同或不同;C1、C2、C3的羟基取代构型为α构型或β构型;且化合物1α,2α-二羟基齐墩果酸和1α,2α-二羟基熊果酸除外。本发明提供了一组结构新颖的化合物及其合成方法,化合物及其盐具有显著的肝损伤保护作用,可用于保肝药物的制备;合成方法反应条件温和,无需特殊仪器设备,操作简单易行,易于扩大规模、实现工业化生产的目的。
说明书
技术领域技术领域
本发明涉及1,2,3-三羟基取代五环三萜类化合物及其制备方法和应用,属于药物化学技术领域。
技术背景背景技术
肝细胞损伤是各型肝病共同的病理基础,其中包括病毒、酒精和化学物质三大因素所引起的肝损伤。肝损伤性疾病已成常见病、多发病,发病率越来越高。我国是肝病的高发地区,因此研究保肝药物意义重大。
当前,我国虽然在临床上广泛应用甘草甜素、联苯双酯、当飞利肝宁和黄芪注射液等保肝、抗肝损伤中药有效成分或复方,但经国际认可的该类药物均不是我国自主发明的(刘平,理论联系实际,基础结合临床,促进中西医结合肝脏病学科发展,中西压结合学报,2003,1(2):81-83)。因此,如何从传统中药中挖掘具有自主知识产权的保肝类药物,一直是我们努力的目标。
普遍存在于植物中的五环三萜化合物齐墩果酸对四氯化碳诱导的肝损伤有保护活性(Liu J.Pharmacology of oleanolic acid and ursolic acid.J Ethnopharmacol1995;49:57-68.);熊果酸对乙醇、硫代乙酰胺、氨基半乳糖和CCL4诱导的小鼠肝损伤有保护作用(Liu J.Pharmacology of oleanolic acid and ursolic acid.JEthnopharmacol 1995;49:57-68;Jeong HG.Inhibition of cytochrome P450 2E1expression by oleanolic acid:Hepatoprotective effects against carbontetrachloride-induced hepatic injury.Toxicol Lett 1999;105:215-222;ShuklaB,Visen PKS,PatnaikGK,Tripathi SC,SrimalRC,DayalR,DobhalPC.Hepatoprotective activity in rat of ursolic acid isolated from Eucalyptushybrid.Phytother Res 1992;6(2):74-79.)。本发明人在开展熊果酸和齐墩果酸的结构衍生物及生物活性筛选研究(白育军,杨小生,康文艺等,熊果酸的结构修饰物及其抗肿瘤活性,华西药学杂志,2003,18(2):87~90;陈磊,杨小生,杨娟等,五环三萜衍生物的合成和对α-葡萄糖苷酶的抑制活性,中国药科大学学报,2010,41(3):222-225)时发现,五环三萜类化合物的A环多氧特别是1,2,3-三羟基取代物具有较强的保肝活性。
因此,本发明对熊果酸和齐墩果酸类化合物进行结构改造,以期发现高保肝活性的A环不同三羟基取代构型的衍生物。
发明内容发明内容
本发明的目的在于:提供具有保肝活性的三羟基取代五环三萜类化合物及其制备方法和应用。本发明将对熊果酸和齐墩果酸类化合物进行结构改造后的系列化合物进行了CCl4所致肝细胞损伤的保护活性筛选,发现该类化合物对人肝细胞具有明显的保护作用,具有制备保肝药物的用途。
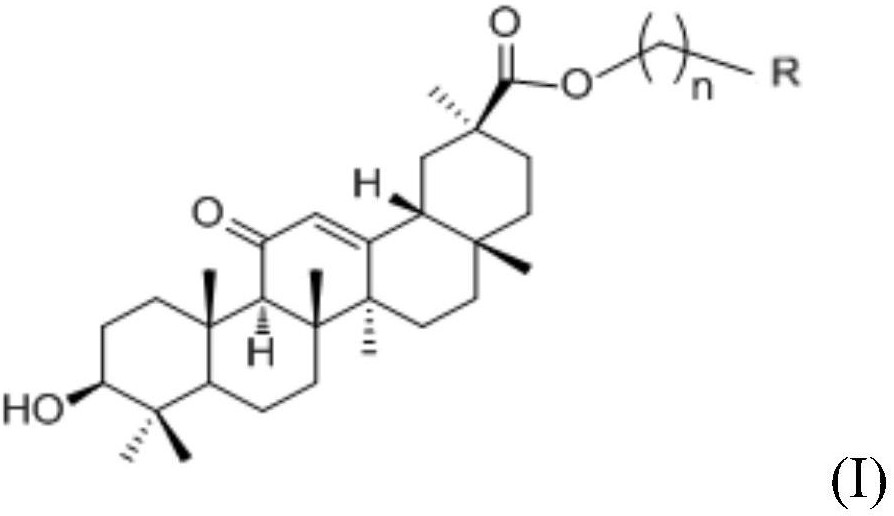
本发明的技术方案:1,2,3-三羟基取代五环三萜类化合物,其通式为下述结构式I:
其中:R1代表氢或甲基;R2代表氢或甲基;R3选自-COOH、-CH2OH、CHO、-COOR1、-CONH2、-CONHR1、-CONR1R2中任一种;R1选自含有1-8个碳原子的烷基、取代或未取代的苯基、取代或未取代的苯烷基中的一种,R2选自含有1-8个碳原子的烷基、取代或未取代的苯基、取代或未取代的苯烷基中的一种,R2与R1可相同或不同;C1的羟基取代构型为α构型或β构型,C2的羟基取代构型为α构型或β构型,C3的羟基取代构型为α构型或β构型;且化合物1α,2α-二羟基齐墩果酸和1α,2α-二羟基熊果酸除外。
前述化合物为齐墩果烷型,即R1为氢,R2为甲基,R3为-COOH;或者为熊果烷型,即R1为甲基,R2为氢,R3为-COOH。
前述化合物优选为以下十四种:(1)1α,2α,3α-三羟基齐墩果-12-烯-28-酸;(2)1α,2β,3α-三羟基齐墩果-12-烯-28-酸;(3)1β,2α,3α-三羟基齐墩果-12烯-28-酸;(4)1β,2β-二羟基齐墩果酸;(5)1α,2β-二羟基齐墩果酸;(6)1β,2α-二羟基齐墩果酸;(7)1β,2β,3α-三羟基齐墩果-12-烯-28-酸;(8)1α,2α,3α-三羟基熊果-12-烯-28-酸;(9)1α,2β,3α-三羟基熊果-12-烯-28-酸;(10)1β,2α,3α-三羟基熊果-12-烯-28-酸;(11)1β,2β-二羟基熊果酸;(12)1α,2β-二羟基熊果酸;(13)1β,2α-二羟基熊果酸;(14)1β,2β,3α-三羟基熊果-12-烯-28-酸;其结构式分别为:
前述化合物进一步优选为:(1)1α,2α,3α-三羟基齐墩果-12-烯-28-酸(简称A-5);(2)1α,2β,3α-三羟基齐墩果-12-烯-28-酸(简称A-6);(8)1α,2α,3α-三羟基熊果-12-烯-28-酸(简称U-17);(9)1α,2β,3α-三羟基熊果-12-烯-28-酸(简称U-18)。
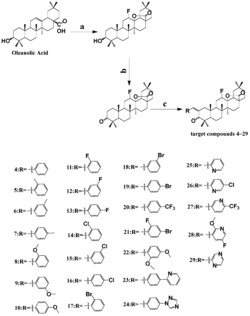
前述的1,2,3-三羟基取代五环三萜类化合物的制备方法之一:取熊果酸U-1,经Jones试剂(将26.72克三氧化铬溶于23毫升浓硫酸中,然后以水稀释至100毫升所得的水溶液)氧化得化合物U-2,U-2经苄溴(即溴化苄)苄基化得化合物U-3,U-3经SeO2氧化得α,β不饱和酮Se-1,Se-1在过氧化氢的作用下得U-15,硼氢化钠还原U-15得R-10-2中间体,再经高氯酸水解开环得FU-16,钯碳催化氢化后得不同异构体产物(8)~(14);其反应路线如下:
制备方法一的具体过程为:①取1摩尔份的熊果酸,用丙酮溶解,置于冰水浴中搅拌15min,缓慢滴加1.14摩尔份的Jones试剂,在N2保护下搅拌反应5h(TLC检测反应进程),用NaOH水溶液调节pH至中性,减压蒸去溶剂,用乙酸乙酯萃取,所得残余物经硅胶柱层析纯化得U-2;②取1摩尔份的U-2,加入丙酮溶解,再加10摩尔份的K2CO3,室温下搅拌30min,再加1.17摩尔份的溴化苄,在N2保护下反应18h(TLC检测反应进程),减压除去丙酮,用乙酸乙酯萃取,有机层用饱和食盐水洗涤、无水MgSO4干燥,减压除去溶剂,残留物经硅胶柱层析得U-3;③取1摩尔份的U-3和1.5摩尔份的SeO2,加入乙酸酐,在N2保护下回流反应5h(TLC检测反应进程),用2N氢氧化钠水溶液终止反应,加入适量水后用乙酸乙酯萃取,有机层用饱和氯化钠水溶液洗涤、无水MgSO4干燥,减压除去溶剂,残余物经硅胶柱层析得Se-1;④取1摩尔份Se-1,加入甲醇溶解,再加入10%NaOH水溶液(含NaOH1.5摩尔份),搅拌30min后,加入30%H2O2(10摩尔份),在N2保护下回流反应3h(TLC检测反应进程),用盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得U-15;⑤取1摩尔份U-15,加入甲醇溶解,在0~5℃下搅拌15min后,分2~3次加入10摩尔份NaBH4,在N2保护下反应2h(TLC检测反应进程),用盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得R-10-2;⑥取1摩尔份R-10-2,加入丙酮溶解,加入1.5摩尔份高氯酸,再加入蒸馏水两滴,在N2保护下反应18h(TLC检测反应进程),用氢氧化钠水溶液调节pH至中性,回收丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得FU-16;⑦取FU-16溶解于无水甲醇中,加入0.1倍于FU-16重量的钯碳作为催化剂,室温下氢化还原,即得U-18及其异构体产物[化合物(8)~(14)]。
前述的1,2,3-三羟基取代五环三萜类化合物的制备方法之二:取熊果酸U-1,经Jones试剂氧化得化合物U-2,U-2经SeO2氧化得α,β不饱和酮Se,Se在过氧化氢的作用下得U-15-1,硼氢化钠还原U-15-1得R-10-21中间体,再经高氯酸水解开环得不同异构体产物(8)~(14);其反应路线如下:
制备方法二的具体过程为:①取1摩尔份的熊果酸,用丙酮溶解,置于冰水浴中搅拌15min,缓慢滴加1.14摩尔份的Jones试剂,在N2保护下搅拌反应5h(TLC检测反应进程),用NaOH水溶液调节pH至中性,减压蒸去溶剂,用乙酸乙酯萃取,所得残余物经硅胶柱层析纯化得U-2;②取1摩尔份的U-2和2摩尔份的SeO2,加入乙酸酐,在氮气保护下回流反应5h(TLC检测反应进程),用2N氢氧化钠水溶液终止反应,加入适量水后用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得Se;③取1摩尔份的Se,加入甲醇溶解,再加入10%NaOH水溶液(含NaOH1.5摩尔份),搅拌30min后,加入30%H2O2(5摩尔份),在N2保护下回流反应3h(TLC检测反应进程),用盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得U-15-1;④取1摩尔份的U-15-1,加入甲醇溶解,在0~5℃下搅拌15min后,分2~3次加入5摩尔份的NaBH4,在N2保护下反应2h(TLC检测反应进程),用盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得R-10-21;⑤取1摩尔份的R-10-21,加入丙酮溶解,加入1.5摩尔份的高氯酸,再加入蒸馏水一滴,在N2保护下反应18h(TLC检测反应进程),用氢氧化钠水溶液调节pH至中性,回收丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂后,残余物经硅胶柱层析得U-18及其异构体产物[化合物(8)~(14)];同样分离得到量少、在硫酸显色剂下呈红色的样品U-17,由EI-MS和1H-NMR确定其结构为1α,2α,3α-三羟基熊果-12-烯-28-酸。
前述的1,2,3-三羟基取代五环三萜类化合物的制备方法之三:取熊果酸或其甲酯或其苄基酯U-a在甲基磺酰氯作用下得U-b,U-b经碳酸锂处理,得中间体U-c,U-c通过二氧化硒氧化得U-d,再经NaOH水溶液处理后脱羧得U-e,中间体U-e在间氯过氧苯甲酸作用下得环氧化中间体U-f,U-f经高氯酸水解后得1,2,3-三羟基中间体U-g,采用钯碳催化氢化或脱甲基反应后得不同异构体的1,2,3-三羟基熊果-12-烯-28酸产物,即化合物(8)~(14);其反应路线如下:
制备方法三的具体过程为:①取1摩尔份的熊果酸或其甲酯或苄基酯(U-a)溶于吡啶溶剂中,冰水浴中搅拌下,缓慢滴加1.2摩尔份的甲基磺酰氯,在N2保护下搅拌反应5h(TLC检测反应进程),用乙酸乙酯萃取,2N的盐酸水溶液洗去吡啶,所得残余物经硅胶柱层析纯化得U-b;②取1摩尔份的U-b,用N,N’-二甲基甲酰胺溶解,加入碳酸锂2摩尔份,加热回流1小时(TLC检测反应进程),冷却至室温,滤除碳酸锂,加入适量水后,用乙酸乙酯萃取,有机相洗至中性,减压回收溶剂,经硅胶柱层析纯化(石油醚∶乙酸乙酯为洗脱剂),得中间体U-c;③取1摩尔份的U-c和2摩尔份二氧化硒(SeO2),加入乙酸溶解,回流5小时(TLC检测反应进程),冷却至室温,滤除二氧化硒,饱和碳酸氢钠中和至中性,加入适量水后,用乙酸乙酯萃取,减压回收溶剂,残留物经硅胶柱层析纯化(石油醚∶乙酸乙酯为洗脱剂),得中间体U-d;④取1摩尔份的U-d,加入甲醇溶解,再加入10%NaOH(2摩尔份)水溶液,回流1小时,用2N HCl调PH值至中性,乙酸乙酯萃取,回收溶剂后所得残留物经柱层析分离,得白色粉末U-e;⑤将1摩尔份的U-e溶解在二氯甲烷中,加入1.2摩尔份间氯过氧苯甲酸,室温下,在N2保护下反应过夜(TLC检测反应进程),用氯仿萃取,回收溶剂后残留物进行硅胶柱层析分离纯化,得白色粉末U-f;⑥取1摩尔份的U-f,加入丙酮溶解,加入1.5摩尔份的高氯酸,再加入蒸馏水一滴,在N2保护下反应18h(TLC检测反应进程),用氢氧化钠水溶液调节pH至中性,回收丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂后,残余物经硅胶柱层析得U-g;⑦取1摩尔份U-g(R为甲基),加入二甲基甲酰胺溶解,再加入1.2摩尔份碘化锂,回流5小时,冷却至室温后,加入适量水,用乙酸乙酯萃取,回收溶剂后残留物经硅胶柱层析分离纯化,得相应的不同异构体的1,2,3-三羟基熊果-12-烯-28酸产物,即化合物(8)~(14);如果U-g中的R为苄基,则采用钯碳催化氢化,同样可得不同异构体的1,2,3-三羟基熊果-12-烯-28酸产物,即化合物(8)~(14)。
前述的1,2,3-三羟基取代五环三萜类化合物的制备方法之四:取齐墩果酸,经Jones试剂氧化成A-1,后经SeO2氧化得α,β不饱和酮A-2,A-2在过氧化氢的作用下得A-3,硼氢化钠还原A-3得A-4中间体,经高氯酸水解开环得不同异构体产物(1)~(7);其反应路线如下:
制备方法四的具体过程为:①取1摩尔份的齐墩果酸,用丙酮溶解,置于冰水浴中,搅拌15min后缓慢滴加1.14摩尔份的Jones试剂,在氮气保护下搅拌反应5h(TLC检测反应进程),用NaOH水溶液调节pH至中性,减压蒸去丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析(石油醚∶乙酸乙酯)得A-1;②取1摩尔份的A-1和2摩尔份的SeO2,加入乙酸酐,在氮气保护下回流反应5h(TLC检测反应进程),用2N氢氧化钠水溶液终止反应,加入适量水后用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压浓缩后的残余物经硅胶柱层析(石油醚∶乙酸乙酯)得A-2;③取1摩尔份的A-2,加入甲醇溶解,加入10%NaOH水溶液(含NaOH1.5摩尔份),搅拌30min后,加入30%H2O2(5摩尔份),在N2保护下回流反应3h(TLC检测反应进程),用盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液和饱和食盐水洗涤,用无水MgSO4干燥,减压回收溶剂后的残余物经硅胶柱层析(石油醚∶乙酸乙酯)得A-3;④取1摩尔份的A-3,加入甲醇溶解,在0~5℃下搅拌15min后,分2~3次加入5摩尔份的NaBH4,在N2保护下反应2h(TLC检测反应进程),用盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂后的残余物经硅胶柱层析(石油醚∶乙酸乙酯)得A-4;⑤取1摩尔份的A-4,加入丙酮溶解,加入1.5摩尔份的高氯酸,再加入蒸馏水一滴,在N2保护下反应18h(TLC检测反应进程),用氢氧化钠水溶液调节pH至中性,回收丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂后,残余物经硅胶柱层析(石油醚∶乙酸乙酯)得A-6及其异构体产物[化合物(1)~(7)];同时分离得到少量、在硫酸显色剂下呈红色的样品A-5,由EI-MS和1H-NMR可以确定其结构为1α,2α,3α-三羟基齐墩果-12-烯-28-酸。
前述的1,2,3-三羟基取代五环三萜类化合物的制备方法之五:取齐墩果酸或其甲酯或其苄基酯A-a在甲基磺酰氯作用下得A-b,A-b经碳酸锂处理,得中间体A-c,A-c通过二氧化硒氧化得A-d,再经NaOH水溶液处理后脱羧得A-e,中间体A-e在间氯过氧苯甲酸作用下得环氧化中间体A-f,A-f经高氯酸水解后得1,2,3-三羟基中间体A-g,采用钯碳催化氢化或脱甲基反应后得不同异构体的1,2,3-三羟基齐墩果-12-烯-28酸产物,即化合物(1)~(7);其反应路线如下:
制备方法五的具体过程为:①取1摩尔份的齐墩果酸或其甲酯或苄基酯(A-a)溶于吡啶溶剂中,冰水浴中搅拌下,缓慢滴加1.2摩尔份的甲基磺酰氯,在N2保护下搅拌反应5h(TLC检测反应进程),用乙酸乙酯萃取,2N的盐酸水溶液洗去吡啶,所得残余物经硅胶柱层析纯化得A-b;②取1摩尔份的A-b,用N,N’-二甲基甲酰胺溶解,加入碳酸锂2摩尔份,加热回流1小时(TLC检测反应进程),冷却至室温,滤除碳酸锂,加入适量水后,用乙酸乙酯萃取,有机相洗至中性,减压回收溶剂,经硅胶柱层析纯化(石油醚∶乙酸乙酯为洗脱剂),得中间体A-c;③取1摩尔份的A-c,加入乙酸溶解,再加入2摩尔份二氧化硒(SeO2),回流5小时(TLC检测反应进程),冷却至室温,滤除二氧化硒,饱和碳酸氢钠中和至中性,加入适量水后,用乙酸乙酯萃取,减压回收溶剂,残留物经硅胶柱层析纯化(石油醚∶乙酸乙酯为洗脱剂),得中间体A-d;④取1摩尔份的A-d,加入甲醇溶解,再加入10%NaOH(2摩尔份)水溶液,回流1小时,用2N HCl调PH值至中性,乙酸乙酯萃取,回收溶剂后所得残留物经柱层析分离,得白色粉末A-e;⑤将1摩尔份的A-e溶解在二氯甲烷中,加入1.2摩尔份间氯过氧苯甲酸,室温下,在N2保护下反应过夜(TLC检测反应进程),用氯仿萃取,回收溶剂后残留物进行硅胶柱层析分离纯化,得白色粉末A-f;⑥取1摩尔份的A-f,加入丙酮溶解,加入1.5摩尔份的高氯酸,再加入蒸馏水一滴,在N2保护下反应18h(TLC检测反应进程),用氢氧化钠水溶液调节pH至中性,回收丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂后,残余物经硅胶柱层析得A-g;⑦取1摩尔份A-g(R为甲基),加入二甲基甲酰胺溶解,再加入1.2摩尔份碘化锂,回流5小时,冷却至室温后,加入适量水,用乙酸乙酯萃取,回收溶剂后残留物经硅胶柱层析分离纯化,得相应的不同异构体的1,2,3-三羟基齐墩果-12-烯-28酸产物,即化合物(1)~(7);如果A-g中R为苄基,则采用钯碳催化氢化,同样可得不同异构体的1,2,3-三羟基齐墩果-12-烯-28酸产物,即化合物(1)~(7)。
前述的1,2,3-三羟基取代五环三萜类化合物(通式I)中R3为-CH2OH的相应化合物的制备方法为:以化合物(1)~(14)为原料,在LiAlH4作用下发生还原反应,即得;其反应路线如下:
该方法的具体过程为:取1摩尔份的化合物(1)~(14),溶解在无水四氢呋喃或无水乙醚溶剂中,分2~3次加入LiAlH4(5摩尔份),在室温下反应10小时或回流5小时(TLC检测反应进程);反应完毕后,用饱和氯化铵水溶液终止反应,用2N的盐酸中和反应液后,用乙酸乙酯萃取,回收溶剂后残留物经硅胶柱层析分离纯化,得相应的通式I中R3为-CH2OH的1,2,3-三羟基取代五环三萜类化合物。
前述的1,2,3-三羟基取代五环三萜类化合物(通式I)中R3为-CHO的相应化合物的制备方法为:以化合物(1)~(14)为原料,与二异丁基氢化铝(i-Bu2AlH)反应,即得;其反应路线如下:
该方法的具体过程为:取1摩尔份的化合物(1)~(14),溶解在无水四氢呋喃或无水乙醚溶剂中,分2~3次加入二异丁基氢化铝(i-Bu2AlH)(5摩尔份),氮气保护下,室温下反应12小时(TLC检测反应进程);反应完毕后,用饱和氯化铵水溶液终止反应,用2N的盐酸中和反应液后,用乙酸乙酯萃取,回收溶剂后残留物经硅胶柱层析分离纯化,得相应的通式I中R3为-CHO的1,2,3-三羟基取代五环三萜类化合物。
前述的1,2,3-三羟基取代五环三萜类化合物(通式I)中R3为-COOR1、-CONH2、-CONHR1或-CONR1R2的相应化合物的制备方法为:以化合物(1)~(14)为原料,在脱水剂DCC(即N,N-二环己基碳二亚胺)或WSC作用下,与相应的醇R1OH或胺R1NH2、R1R2NH、BnNH2反应,得对应的酯类或酰胺类化合物,即通式I中R3为-COOR1、-CONH2、-CONHR1或-CONR1R2的相应化合物;其反应路线如下:
上述方法的具体过程为:
取1摩尔份的化合物(1)~(14)和1.5摩尔份的醇R1OH或胺R1NH2、R1R2NH,溶解在无水DMF(二甲基甲酰胺)溶剂中,加入3摩尔份的脱水剂DCC或WSC以及0.1摩尔份的DMAP(4-二甲氨基吡啶),氮气保护下,室温反应24小时(TLC检测反应进程),加入2N的盐酸水溶液终止反应后,用乙酸乙酯萃取,回收溶剂后残留物经硅胶柱层析分离纯化,得相应的通式I中R3为-COOR1、-CONHR1或-CONR1R2的1,2,3-三羟基取代五环三萜类化合物。
取1摩尔份的化合物(1)~(14)和1.5摩尔份的苄胺(BnNH2),溶解在无水1,4-二氧六环溶剂中,加入3摩尔份的脱水剂DCC或WSC以及0.1摩尔份的DMAP(4-二甲氨基吡啶),氮气保护下,室温反应24小时(TLC检测反应进程),加入2N的盐酸水溶液终止反应后,用乙酸乙酯萃取,回收溶剂后残留物经硅胶柱层析分离纯化,所得物溶于甲醇中,经钯碳催化氢化还原12小时(室温下),得相应的通式I中R3为-CONH2的1,2,3-三羟基取代五环三萜类化合物。
前述的1,2,3-三羟基取代五环三萜类化合物在制备保肝药物中的应用:将所述化合物或者所述化合物溶解在氢氧化钠、氢氧化钾或氨水溶液中制得的可药用盐与适当辅料制成保肝药物制剂。
前述药物制剂为硬胶囊剂、片剂、口服液、软胶囊剂、颗粒剂或滴丸剂。
本发明所提供的几种制备方法各有优势,可根据需要选择。其中制备方法三、五是对制备方法一、二、四的补充,可高收率获得通过方法一、二、四合成的含量较少的1,2,3-三羟基异构体。按照本发明所提供的制备方法,可有效合成1,2,3-三羟基不同取代构型的目标产物;且本发明提供的方法反应条件温和,无需特殊试剂和严格的反应条件,操作简单易行,易于扩大规模、实现工业化生产的目的。
本发明所述1,2,3-三羟基取代五环三萜类化合物的应用方法,是将所述化合物作为药物来应用:可以加入惰性辅助剂和/或赋形剂将所述化合物制备成适宜的给药剂型;也可以将所述化合物作为肝损伤保护剂用于肝病的防治药物中。药物可以是片剂、胶囊剂、注射剂或丸剂等。具体应用方法可以采用现有技术中的常规方法,即按照本领域公知的制备方法,采用传统的添加剂、赋形剂制备;可以将一种或几种活性物质与一种或几种添加剂、赋形剂混合形成药用组合物,再将组合物制成药物。由此制得的药物根据需要可以非肠道、口服等途径给药。
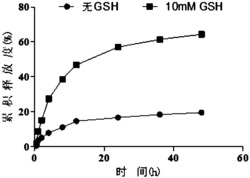
经过试验研究发现,本发明所述1,2,3-三羟基取代五环三萜类化合物对四氯化碳诱导肝细胞损伤具有很好的保护作用,可以作为保肝药物应用。以下是为验证本发明所述化合物的生物活性效果而进行的生物活性试验。
1、体外对CCl4所致肝细胞损伤的保护作用
试验样品:A-5(即1α,2α,3α-三羟基齐墩果-12-烯-28-酸)及其钠盐;A-6(即1α,2β,3α-三羟基齐墩果-12-烯-28-酸)及其钠盐;U-17(即1α,2α,3α-三羟基熊果-12-烯-28-酸)及其钠盐;U-18(即1α,2β,3α-三羟基熊果-12-烯-28-酸)及其钠盐;AM(1,2,3-三羟基齐墩果-12-烯-28-酸、即化合物(1)~(7)的混合物)及其钠盐;UM(1,2,3-三羟基熊果-12-烯-28-酸、即化合物(8)~(14)的混合物)及其钠盐;FU-16(实施例6制得);A-6-CH2OH(以A-6为原料制得,见实施例31);A-6-CHO(以A-6为原料制得,见实施例32);A-6-CONHBn(以A-6为原料制得,见实施例34);A-6-CONH2(以A-6为原料制得,见实施例35)。
方法:常规体外培养人肝HL-7702细胞接种于96孔板,分别给予不同浓度(见表1、表2和表3)的17种样品继续培养12h后,用三种不同剂量(8μl、6μl、4μl)CCl4造成肝损伤,每一种药物均设两个平行对照孔。应用MTS法在酶联免疫检测仪490nm测定吸光度,观察药物对CCl4所致肝细胞毒性的保护作用。相对正常组计算细胞生存率,用spss软件进行单因变量多因素方差分析。
结果:CCl4模型组对肝细胞造成明显损伤,而应用17种样品药物的肝细胞生存率与模型组细胞生存率具有显著性差异,与正常组细胞生存率没有明显差异(见表l、表2和表3)。
结论:所有试验样品(药物)均能显著提高模型组细胞的生存率,对人肝HL-7702细胞具有明显的保护作用。
试验结果:
表l体外对CCl4所致肝细胞损伤的保护作用(8μl CCl4)
*给药组vs模型组,p<0.01;#模型组vs正常组,p<0.01。
表2体外对CCl4所致肝细胞损伤的保护作用(6μl CCl4)
*给药组vs模型组,p<0.01;#模型组vs正常组,p<0.01。
表3体外对CCl4所致肝细胞损伤的保护作用(4μl CCl4)
*给药组vs模型组,p<0.01;#模型组vs正常组,p<0.01。
2、体内对CCl4所致小鼠肝细胞损伤的保护作用
A、试验材料
(1)受试药物(样品)
A-6(即1α,2β,3α-三羟基齐墩果-12-烯-28-酸)及其钠盐;U-18(即1α,2β,3α-三羟基熊果-12-烯-28-酸);AM(1,2,3-三羟基齐墩果-12-烯-28-酸、即化合物(1)~(7)的混合物);UM(1,2,3-三羟基熊果-12-烯-28-酸、即化合物(8)~(14)的混合物)及其钠盐。
以上样品(药物)实验前用0.5%CMC配制成所需浓度混悬液。
(2)药品及试剂
阳性药:联苯双酯滴丸浙江医药股份有限公司新昌制药厂批号:091206。
双环醇北京协和药厂批号:100602。
B、试验动物
(1)来源及种属:KM小鼠,雌雄兼用,体重18~22g。由贵阳医学院实验动物中心提供,动物合格证号:SCXK(黔)2002-0001。
(2)饲养条件:KM小鼠,雌雄分笼饲养于洁净动物饲养柜中,小鼠每笼10只。动物室光照充足,通风和空调设备良好,室温18~25℃,相对湿度50~70%。
(3)数据分析与处理
数据以均数标准差 表示,采用Excel 2000作统计学处理,计量资料采用Student-t检验。
C、给药途径
根据临床用药途径,试验动物采用灌胃给药。
D、试验方法与结果
(1)试验方法:
取KM小鼠36只,雌雄各半,体重18~22g,随机分为6组:模型组(等容积0.5%CMC)、正常组、联苯双酯组(3.75mg/kg)、双环醇组(12.5mg/kg)、试验样品低剂量组(50mg/kg)、试验样品高剂量组(170mg/kg),每组6只。阳性药为市售联苯双酯滴丸与双环醇片。给药容积20ml/kg,每天2次,连续给药2天。各组末次给药8h后腹腔注射0.1%CCl4花生油10ml·kg-1。禁食16h后处死小鼠,取血离心做ALT检查。
(2)试验结果(见表4):
表4对四氯化碳所致小鼠肝损伤的保护作用
与模型组比较,*P<0.05,**P<0.01。
结果表明,所有试验样品(药物)高、低剂量组均可明显降低CCl4中毒小鼠的ALT,与模型组比较差异显著(P<0.01,P<0.05),其中试验样品的低剂量组与模型组比较有极显著性差异(P<0.01),比阳性药效果还要明显。
综上所述,本发明提供的1,2,3-三羟基取代五环三萜类化合物对四氯化碳诱导的肝(细胞)损伤有显著的保护作用,预期可作为保肝药物应用。
与现有技术相比,本发明提供了一组结构新颖的1,2,3-三羟基取代的五环三萜衍生物类化合物及其合成方法,所述合成方法反应条件温和,无需特殊仪器设备,操作简单易行,易于扩大规模、实现工业化生产的目的;且本发明所提供的化合物及其可药用盐(钠盐、钾盐或铵盐)具有显著的肝损伤保护作用(体内和体外),可用于保肝药物的制备。
附图说明具体实施方式具体实施方式
实施例1:化合物U-2的合成
取1.000g熊果酸(2.19mmol)(即U-1)于250mL圆底烧瓶中,用120mL丙酮溶解,置于冰水浴中搅拌15min,缓慢滴加Jones试剂0.936mL(2.5mmol),在N2保护下搅拌5h反应完成(TLC检测反应进程)。用2mol·L-1NaOH水溶液调节pH至中性,减压蒸去溶剂,用乙酸乙酯萃取,所得残余物经硅胶柱层析纯化得946mg U-2(95.0%)。
U-2白色粉末,mp 256~258℃;IR(KBr)υmax1694cm-1,2922cm-1;1H-NMR(CDCl3)δ:5.25(1H,s,H-12),1.08,1.08,1.05,1.02,0.82(3H×5,s,23,24,25,26,27),0.96~0.95(3H,d,J=4.0Hz,H-29),0.87~0.85(3H,d,J=6.0Hz,H-30);13C-NMR(CDCl3)δ:217.8(C-3),183.8(C-28),138.0(C-13),125.5(C-12),55.2(C-5),52.5(C-18),47.9(C-17),47.3(C-8),46.7(C-9),42.0(C-14),39.4(C-4),39.2(C-1),39.0(C-20),38.7(C-19),36.6(C-22),36.6(C-10),34.1(C-7),32.4(C-21),30.5(C-15),27.9(C-12),26.5(C-23),24.0(C-16),23.5(C-27),23.3(C-11),21.4(C-30),31.1(C-29),19.5(C-6),16.9(C-25),16.9(C-26),15(C-24);MS(EI)m/z:454(M+),248(100),203,133,55,43。
实施例2:化合物U-3的合成
取414mg(0.912mmol)U-2于250mL的圆底烧瓶中,加入25mL的丙酮溶解,再加1.2g(9.12mmol)K2CO3,室温下搅拌30min,再加128μL(1.07mmol)溴化苄,在N2保护下反应18h反应完成(TLC检测反应进程),减压除去丙酮,用乙酸乙酯萃取,有机层用饱和食盐水洗涤、无水MgSO4干燥,减压除去溶剂,残留物经硅胶柱层析得430mg U-3(86.7%)。
U-3白色粉末,mp 96~98℃;IR(KBr)υmax1715cm-1,2940cm-1,1450cm-1,1379cm-1;1H-NMR(CDCl3)δ:7.35(s,5H),5.28(1H,s,H-12),4.97-5.12(2H,dd,benzyl),1.08,1.03,1.02,1.00,0.68(3H×5,s,23,24,25,26,27),0.94~0.92(3H,d,J=9.2Hz,H-29),0.86~0.84(3H,d,J=6.8Hz,H-29);13C-NMR(CDCl3)δ:217.8(C-3),177.1(C-28),138.1(C-13),136.2(s),128.4(s),128.2(s),128.1(s),127.9(s),127.9(s),125.4(C-12),65.9(benzyl),55.2(C-5),52.9(C-18),48.0(C-17),47.3(C-8),46.7(C-9),42.0(C-14),39.4(C-4),39.2(C-1),39.0(C-20),38.7(C-19),36.5(C-22),36.5(C-10),34.1(C-7),32.4(C-21),30.6(C-15),27.8(C-12),26.5(C-23),24.1(C-16),23.4(C-27),23.3(C-11),21.4(C-30),21.1(C-29),19.5(C-6),16.9(C-25),16.9(C-26),15.1(C-24);MS(EI)m/z:544(M+),91(100),248,203,133,55,43。
实施例3:化合物Se-1的合成
取400mg(0.735mmol)U-3、110mg(1.10mmol)SeO2于50mL圆底烧瓶中,加入20mL乙酸酐,在N2保护下回流5h反应完成(TLC检测反应进程),用2mol·L-1氢氧化钠水溶液终止反应,加入适量水后用乙酸乙酯萃取,有机层用饱和氯化钠水溶液洗涤、无水MgSO4干燥,减压除去溶剂,残余物经硅胶柱层析得191mg Se-1(47.9%)。
Se-1白色粉末,mp 127~130℃;IR(KBr)υmax1724cm-1,2926cm-1,1667cm-1,1456cm-1,1383cm-1;1H-NMR(CDCl3)δ:7.35(s,5H),7.06~7.04(d,J=10.4,1H,H-1),5.81~5.79(1H,d,J=10.4Hz,H-2),5.29(1H,s,H-12),4.98-5.13(2H,dd,benzyl);2.31~2.28(1H,d,J=11.2,H-18),1.14,1.13,1.09,1.03,0.70(3H×5,s,23,24,25,26,27),0.94~0.93(3H,d,J=6.8Hz,H-29),0.87~0.85(3H,d,J=6.4Hz,H-30)ppm;13C-NMR(CDCl3)δ:205.4(C-3),177.2(C-28),159.4(C-1),138.6(C-13),136.2(s),128.3(s),128.2(s),128.0(s),127.9(s),127.9(s),124.9(C-2),124.9(C-12),65.9(benzyl),53.3(C-5),53.0(C-18),48.1(C-17),44.5(C-14),42.3(C-4),41.6(C-9),40.2(C-8),39.2(C-10),38.9(C-20),38.7(C-19),36.5(C-22),32.7(C-7),30.6(C-21),29.6(C-15),24.1(C-16),23.3(C-23),23.1(C-11),21.6(C-27),21.0(C-30),18.8(C-6),18.7(C-26),17.5(C-25),16.9(C-24);MS(EI)m/z 452(M+),248(100),203,133。
实施例4:化合物U-15的合成
取200mg(0.442mmo)Se-1于50mL圆底烧瓶中,加入25mL甲醇溶解,加入10%NaOH水溶液265μL(0.663mmol),搅拌30min后,加入500μL(4.42mmol)30%H2O2,在N2保护下回流3h反应完成(TLC检测反应进程),用2mol·L-1盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得152mg U-15(74%)。
U-15白色粉末,mp 95~98℃;IR(KBr)υmax1725cm-1,2926cm-1,1694cm-1,1466cm-1,1385cm-1;1H-NMR(CDCl3)δ:7.35(s,5H),5.28(1H,s,H-12),5.13~4.97(2H,dd,benzyl),3.51~3.50(1H,d,J=4.4Hz,H-2),3.38~3.37(1H,d,J=4.4Hz,H-1),2.31~2.28(1H,d,J=11.2Hz,H-18),1.12,1.08,0.99,0.95,0.70(3H×5,s,23,24,25,26,27),0.94~0.93(d,J=4.0Hz,3H,H-29),0.87~0.86(d,J=6.4Hz,3H,H-30)ppm;13C-NMR(CDCl3)δ:212.8(C-3),177.2(C-28),138.7(C-13),136.2(s),128.3(s),128.1(s),128.0(s),127.9(s),127.9(s),124.8(C-12),65.9(C-1),64.0(C-2),56.8(benzyl),53.0(C-18),48.1(C-17),46.7(C-4),45.9(C-5),42.5(C-14),40.4(C-9),39.6(C-8),39.0(C-19),38.7(C-20),38.3(C-10),36.5(C-22),32.4(C-7),30.6(C-21),27.9(C-15),27.9(C-23),24.1(C-16),23.7(C-11),23.5(C-27),23.3(C-30),21.1(C-29),18.7(C-6),17.6(C-26),17.1(C-25),16.9(C-24);MS(EI)m/z 558(M+),91(100),133,467,543。
实施例5:化合物R-10-2的合成
取200mg U-15(0.358mmol)于50mL圆底烧瓶中,加入25mL甲醇溶解,在0~5℃下搅拌15min后,分3次加入136mg(3.58mmol)NaBH4,在N2保护下反应2h反应完成(TLC检测反应进程),用2mol·L-1盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得144mg R-10-2(72%)。
R-10-2白色粉末,mp 101~103℃;IR(KBr)υmax1722cm-1,2925cm-1,1662cm-1,1450cm-1,1384cm-1;1H-NMR(CDCl3)δ:7.35(s,5H),5.25(1H,s,H-12),5.12~4.96(2H,dd,benzyl),3.55(1H,s,H-3),3.07~3.06(1H,d,J=4.0Hz,H-2),2.99~2.98(1H,d,J=3.6Hz,H-1);2.28~2.26(1H,d,J=11.6Hz,H-18),1.07,0.93,0.93,0.78,0.64(3H×5,s,23,24,25,26,27),1.06~1.03(3H,d,J=10.4,H-29),0.87~0.84(3H,d,J=6.4,H-30)ppm;13C-NMR(CDCl3)δ:177.4(C-28),138.3(C-13),136.2(s),128.3(s),128.0(s),127.9(s),127.8(s),127.8(s),124.9(C-12),75.7(C-3),65.9(benzyl),60.3(C-1),57.3(C-2),52.8(C-18),48.0(C-17),45.5(C-5),41.3(C-14),41.0(C-9),39.4(C-8),39.2(C-19),38.9(C-4),38.7(C-20),36.9(C-10),36.5(C-22),32.6(C-7),30.5(C-21),28.5(C-23),27.8(C-15),24.1(C-16),23.4(C-27),3.3(C-11),21.1(C-30),18.1(C-29),17.0(C-26),16.9(C-25),16.7(C-6),16.4(C-24);MS(EI)m/z 560(M+),91(100),247,433,451,524,542。
实施例6:化合物FU-16的合成
取100mg(0.179mmol)R-10-2于50mL圆底烧瓶中,加入25mL丙酮溶解,加入16μL(0.268mmol)高氯酸,再加入蒸馏水两滴,在N2保护下反应18h反应完成(TLC检测反应进程),用2mol·L-1氢氧化钠水溶液调节pH至中性,回收丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得78mg FU-16(76.1%)。
FU-16白色粉末,mp 108~110℃;IR(KBr)υmax3443cm-1,2925cm-1,1627cm-1,1384cm-1;1H-NMR(CD3OD)δ:5.24(1H,s,H-12),5.12~4.96(2H,dd,benzyl),3.93(1H,d,J=6.8Hz,H-2),3.61(1H,d,J=2.4Hz,H-3),3.45(1H,J=d,4.4Hz,H-1);13C-NMR(CD3OD)δ:180.5(C-28),139.5(C-13),136.2(s),128.3(s),128.0(s),127.9(s),127.8(s),127.8(s),127.1(C-12),77.4(C-3),75.3(C-1),74.9(C-2),65.9(benzyl),49.6(C-5),47.6(C-17),47.2(C-19),43.4(C-14),42.7(C-18),41.5(C-8),40.3(C-4),39.7(C-9),39.3(C-20),39.2(C-10),34.8(C-21),33.8(C-7),33.6(C-29),33.5(C-22),30.1(C-30),28.8(C-15),26.5(C-27),24.4(C-16),24.0(C-30),24.0(C-11),19.1(C-6),18.1(C-25),17.8(C-24),14.8(C-26);MS(EI)m/z:578(M+),91(100),248,203,469,524,542,556。
实施例7:化合物U-18及其异构体的合成
取100mg(0.173mmol)FU-16溶解于20mL无水甲醇中,加入10mg钯碳作为催化剂,室温下氢化还原,即得80mgU-18(95%)及其异构体产物[化合物(8)~(14)]。
U-18白色粉末,mp 272~275℃;IR(KBr)υmax3454cm-1,2924cm-1,1682cm-1;1H-NMR(CD3OD)δ:5.24(1H,s,H-12),3.93(1H,d,J=6.8Hz),3.61(1H,d,J=2.4Hz),3.45(1H,d,J=4.4Hz),1.28,1.23,1.13,1.00,0.87(3H×5,s,23,24,25,26,27),0.96~0.94(3H,d,J=6.8Hz,H-29),0.90~0.89(3H,d,J=5.2Hz,H-30);13C-NMR(CD3OD)δ:180.5(C-28),139.5(C-13),127.1(C-12),77.4(C-3),75.3(C-1),74.9(C-2),49.6(C-5),47.6(C-17),47.2(C-19),43.4(C-14),42.7(C-18),41.5(C-8),40.3(C-4),39.7(C-9),39.3(C-20),39.2(C-10),34.8(C-21),33.8(C-7),33.6(C-29),33.5(C-22),30.1(C-30),28.8(C-15),26.5(C-27),24.4(C-16),24.0(C-30),24.0(C-11),19.1(C-6),18.1(C-25),17.8(C-24),14.8(C-26);MS(EI)m/z:488(M+),248(100),43,133,203,452,470.
实施例8:化合物Se的合成
取U-2400mg(0.881mmol)、SeO2176mg(1.76mmol)于50mL圆底烧瓶中,加入20mL乙酸酐,在氮气保护下回流5h反应完成(TLC检测反应进程),用2mol·L-1氢氧化钠水溶液终止反应,加入适量水后用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得191mg Se(46.7%)。
Se白色粉末,mp 127~130℃;IR(KBr)υmax1698cm-1,2962cm-1;1H-NMR(CDCl3)δ:7.07~7.05(1H,d,J=10.4Hz,H-1),5.82~5.80(1H,d,J=10.4Hz,H-2),5.25(1H,s,H-12),2.31~2.28(1H,d,J=11.2Hz,H-18),1.17,1.16,1.10,1.08,0.85(3H×5,s,23,24,25,26,27),0.96~0.94(3H,d,J=6.4Hz,H-29),0.87~0.86(3H,d,J=4.4Hz,H-30)ppm;13C-NMR(CDCl3)δ:205.3(C-3),183.8(C-28),159.3(C-1),138.0(C-13),124.9(C-2),124.9(C-12),53.3(C-5),53.0(C-18),48.1(C-17),44.5(C-14),42.3(C-4),41.6(C-9),40.2(C-8),39.2(C-10),38.9(C-20),38.7(C-19),36.5(C-22),32.7(C-7),30.6(C-21),29.6(C-15),24.1(C-16),23.3(C-23),23.1(C-11),21.6(C-27),21.0(C-30),18.8(C-6),18.7(C-26),17.5(C-25),16.9(C-24);MS(EI)m/z 452(M+),248(100),203,133,437。
实施例9:化合物U-15-1的合成
取400mg Se(0.881mmol)于50mL圆底烧瓶中,加入25mL甲醇溶解,加入10%NaOH水溶液53μL(1.321mmol),搅拌30min后,加入30%H2O2498μL(4.40mmol),在N2保护下回流3h反应完成(TLC检测反应进程),用2mol·L-1盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得317mg U-15-1(76.6%)。
U-15-1白色粉末,mp 138~140℃;IR(KBr)υmax1692cm-1,2923cm-1,1468cm-1;1H-NMR(CDCl3)δ:5.30(1H,s,H-12),3.52~3.51(1H,d,J=4.8,H-2),3.39~3.37(1H,d,J=4.8,H-1),1.25,1.20,1.15,0.96,0.84(3H×5,s,23,24,25,26,27),0.95~0.93(3H,d,J=6.4,H-29),0.86~0.85(3H,d,J=6.0,H-30);13C-NMR(CDCl3)δ:212.7(C-3),183.9(C-28),138.5(C-13),124.9(C-12),63.9(C-1),56.9(C-2),52.6(C-18),48.0(C-17),45.9(C-5),44.7(C-14),42.1(C-20),40.4(C-9),38.9(C-19),38.6(C-20),38.4(C-4),36.7(C-10),36.5(C-22),32.3(C-7),30.5(C-21),28.0(C-15),27.9(C-23),23.9(C-16),23.7(C-11),23.4(C-27),21.1(C-30),20.8(C-29),18.7(C-6),17.6(C-26),16.9(C-25),15.0(C-24);MS(EI)m/z 468(M+),202(100),43,248,422,450。
实施例10:化合物R-10-21的合成
取200mg U-15-1(0.427mmol)于50mL圆底烧瓶中,加入25mL甲醇溶解,在0~5℃下搅拌15min后,分3次加入81mg(2.136mmol)NaBH4,在N2保护下反应2h反应完成(TLC检测反应进程),用2mol·L-1盐酸水溶液调节pH至中性,回收甲醇,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析得139mg R-10-21(69%)。
R-10-21白色粉末,mp 208~210℃;IR(KBr)υmax1691cm-1,2924cm-1,1457cm-1;1H-NMR(CDCl3)δ:5.30(1H,s,H-12),3.49(1H,s,H-3),3.07~3.06(1H,d,J=3.6Hz,H-2),3.02~3.01(1H,d,J=3.6Hz,H-1);2.82~2.65(1H,d,J=9.6Hz,H-18),1.22,1.14,0.93,0.90,0.82,0.80,0.76(3H×7,s,23,24,25,26,27),0.95~0.93(3H,d,J=6.4Hz,H-29),0.86~0.85(3H,d,J=6.0Hz,H-30)ppm;13C-NMR(CDCl3)δ:183.4(C-28),138.3(C-13),124.9(C-12),75.5(C-3),60.3(C-1),57.3(C-2),52.8(C-18),48.0(C-17),45.5(C-5),41.3(C-14),41.0(C-9),39.4(C-8),39.0(C-19),38.9(C-4),38.7(C-20),36.9(C-10),36.5(C-22),32.6(C-7),30.5(C-21),28.5(C-23),27.8(C-15),24.1(C-16),23.4(C-27),23.3(C-11),21.1(C-30),18.1(C-29),17.0(C-26),16.9(C-25),16.7(C-6),16.4(C-24);MS(EI)m/z:470(M+),248(100),43,133,203,452.
实施例11:化合物U-18及其异构体的合成
取100mg R-10-21(0.213mmol)于50mL圆底烧瓶中,加入25mL丙酮溶解,加入18μL(0.319mmol)高氯酸,加入蒸馏水一滴,在N2保护下反应18h反应完成(TLC检测反应进程),用2mol·L-1氢氧化钠水溶液调节pH至中性,回收丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂后,残余物经硅胶柱层析得65mg U-18(62.7%)及其异构体产物[化合物(8)~(14)]。
U-18白色粉末,mp 272~275℃;IR(KBr)υmax3454cm-1,2924cm-1,1682cm-1;1H-NMR(CD3OD)δ:5.24(1H,s,H-12),3.93(1H,d,J=6.8Hz),3.61(1H,d,J=2.4Hz),3.45(1H,d,J=4.4Hz),1.28,1.23,1.13,1.00,0.87(3H×5,s,23,24,25,26,27),0.96~0.94(3H,d,J=6.8Hz,H-29),0.90~0.89(3H,d,J=5.2Hz,H-30);13C-NMR(CD3OD)δ:180.5(C-28),139.5(C-13),127.1(C-12),77.4(C-3),75.3(C-1),74.9(C-2),49.6(C-5),47.6(C-17),47.2(C-19),43.4(C-14),42.7(C-18),41.5(C-8),40.3(C-4),39.7(C-9),39.3(C-20),39.2(C-10),34.8(C-21),33.8(C-7),33.6(C-29),33.5(C-22),30.1(C-30),28.8(C-15),26.5(C-27),24.4(C-16),24.0(C-30),24.0(C-11),19.1(C-6),18.1(C-25),17.8(C-24),14.8(C-26);MS(EI)m/z:488(M+),248(100),43,133,203,452,470.
同样分离得到量少的、在硫酸显色剂下呈红色的样品U-17。由EI-MS和1H-NMR确定其结构为1α,2α,3α-三羟基熊果-12-烯-28-酸。
实施例12:化合物U-b的合成
取1mmol的熊果酸或其甲酯或苄基酯(U-a)溶于吡啶溶剂中,冰水浴中搅拌下,缓慢滴加1.2mmol的甲基磺酰氯,在N2保护下搅拌反应5h(TLC检测反应进程),用乙酸乙酯萃取,2N的盐酸水溶液洗去吡啶,所得残余物经硅胶柱层析纯化得U-b(白色固体粉末)。
实施例13:化合物U-c的合成
取1mmol的U-b,用N,N’-二甲基甲酰胺10毫升溶解,加入碳酸锂2mmol,加热回流1小时(TLC检测反应进程),冷却至室温,滤除碳酸锂,加入适量水后,用乙酸乙酯萃取,有机相洗至中性,减压回收溶剂,经硅胶柱层析纯化(石油醚∶乙酸乙酯混合溶剂为洗脱剂),得中间体U-c(白色固体粉末)。
实施例14:化合物U-d的合成
取1mmol的U-c,加入10毫升乙酸和2mmol二氧化硒(SeO2),回流5小时(TLC检测反应进程),冷却至室温,滤除二氧化硒,饱和碳酸氢钠中和至中性,加入适量水后,用乙酸乙酯萃取,减压回收溶剂,残留物经硅胶柱层析纯化(石油醚∶乙酸乙酯混合溶剂为洗脱剂),得中间体U-d(白色固体粉末)。
实施例15:化合物U-e的合成
取1mmol的U-d,加入10毫升甲醇和5毫升10%NaOH水溶液,回流1小时,用2N HCl调PH值至中性,乙酸乙酯萃取,回收溶剂后所得残留物经柱层析分离,得白色粉末U-e。
实施例16:化合物U-f的合成
将1mmol的U-e溶解在15毫升的二氯甲烷中,加入1.2mmol间氯过氧苯甲酸,室温下,在N2保护下反应过夜(TLC检测反应进程),用氯仿萃取,回收溶剂后残留物进行硅胶柱层析分离纯化,得到白色粉末U-f。
实施例17:化合物U-g的合成
取1mmol的U-f,加入丙酮溶解,加入1.5mmol的高氯酸,再加入蒸馏水一滴,在N2保护下反应18h(TLC检测反应进程),用氢氧化钠水溶液调节pH至中性,回收丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂后,残余物经硅胶柱层析得U-g。
实施例18:1,2,3-三羟基熊果-12-烯-28酸的合成
取1mmolU-g(R为甲基),加入10毫升二甲基甲酰胺和1.2mmol碘化锂,回流5小时,冷却至室温后,加入适量水,用乙酸乙酯萃取,回收溶剂后残留物经硅胶柱层析分离纯化,得相应的不同异构体的1,2,3-三羟基熊果-12-烯-28酸产物,即化合物(8)~(14);如果U-g(R为苄基),则采用钯碳催化氢化,同样可得不同异构体的1,2,3-三羟基熊果-12-烯-28酸产物,即化合物(8)~(14)。
实施例19:化合物A-1的合成
取1.000g齐墩果酸(2.19mmol)于250mL圆底烧瓶中,用120mL丙酮溶解,置于冰水浴中,搅拌15min后缓慢滴加Jones试剂0.936mL(2.5mmol),在氮气保护下搅拌5h反应完成(TLC检测反应进程)。用2mol·L-1NaOH水溶液调节pH至中性,减压蒸去丙酮,残余物加水分散,用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO4干燥,减压回收溶剂,残余物经硅胶柱层析(石油醚∶乙酸乙酯)得0.946g A-1(96.3%)。
A-1白色粉末,mp 128~130℃;IR(KBr)υmax1697cm-1,2924cm-1;1H-NMR(CDCl3)δ:5.30(1H,s,H-12),1.14,1.07,1.04,1.03,0.98,0.93,0.86(3H×7,s,23,24,25,26,27,29,30)ppm;13C-NMR(CDCl3)δ:217.8(C-3),184.4(C-28),143.6(C-13),122.3(C-12),55.2(C-5),47.4(C-17),46.8(C-19),46.5(C-14),45.7(C-8),41.6(C-9),40.9(C-18),39.2(C-4),39.0(C-10),36.7(C-1),34.1(C-22),33.7(C-21),33.0(C-29),32.3(C-7),32.1(C-23),30.6(C-20),27.6(C-15),26.4(C-2),25.8(C-27),23.5(C-30),23.4(C-16),22.8(C-11),21.4(C-26),19.5(C-6),16.9(C-25),15.0(C-24)ppm;MS(EI)m/z 454(M+),203(100),248,43,55,133,439
实施例20:化合物A-2的合成
取A-1400mg(0.881mmol)、SeO2176mg(1.76mmol)于50mL圆底烧瓶中,加入20mL乙酸酐,在氮气保护下回流5h反应完成(TLC检测反应进程),用2mol·L-1氢氧化钠水溶液终止反应,加入适量水后用乙酸乙酯萃取,有机层用饱和NaHCO3水溶液洗涤、饱和食盐水洗涤、无水MgSO
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0