








专利摘要
本发明公开了一种原人参三醇类人参皂苷的制备方法,包括以下步骤:(a)对原人参三醇的羟基进行保护,从而裸露20位羟基并裸露3位、6位和12位中至少一个羟基,得到部分羟基被保护的原人参三醇受体;(b)步骤(a)得到的部分羟基被保护的原人参三醇受体与全保护的糖基给体发生糖苷化反应,得到连接有全保护的糖基、且羟基被保护的原人参三醇类人参皂苷;(c)对步骤(b)得到的连接有全保护的糖基、且羟基被保护的原人参三醇类人参皂苷进行脱保护,从而得到原人参三醇类人参皂苷。本发明的化学合成方法新颖、可靠、高效、收率及立体选择性高。
权利要求
1.一种原人参三醇类人参皂苷的制备方法,其特征在于,所述方法包括以下步骤:
(a)以原人参三醇为原料,对原人参三醇的羟基进行保护,从而裸露20位羟基以及裸露3位、6位和12位中至少一个羟基,得到部分羟基被保护的原人参三醇受体;
(b)步骤(a)得到的部分羟基被保护的原人参三醇受体与全保护的糖基给体发生糖苷化反应,得到连接有全保护的糖基、且部分羟基被保护的原人参三醇类人参皂苷;
(c)对步骤(b)得到的所述连接有全保护的糖基、且部分羟基被保护的原人参三醇类人参皂苷,进行脱保护,从而得到原人参三醇类人参皂苷。
2.如权利要求1所述的制备方法,其特征在于,所述的糖基给体的结构如下所示:
式中,R5为C1-6的烷基、C3-10环烷基或C3-10芳基;
R6为全保护的β-D-葡萄糖基、α-D-葡萄糖基、β-D-半乳糖基、α-D-半乳糖基、β-D-甘露糖基、α-D-甘露糖基、β-D-木糖基、α-D-木糖基、β-D-2-氨基葡萄糖基、α-D-2-氨基葡萄糖基、α-L-鼠李糖基、β-L-鼠李糖基、α-D-阿拉伯糖基、β-D-阿拉伯糖基、α-L-阿拉伯糖基、β-L-阿拉伯糖基、α-L-岩藻糖基、β-L-岩藻糖基、β-D-葡萄糖醛酸基、α-D-葡萄糖醛酸基、β-D-半乳糖醛酸基、或α-D-半乳糖醛酸基;所述全保护是指用乙酰基、苯甲酰基、苄基、或硅基对全部的羟基进行保护。
3.如权利要求1所述的制备方法,其特征在于,所述步骤(a)中以原人参三醇为原料,对原人参三醇的羟基进行保护,从而裸露20位羟基以及裸露3位、6位、20位中至少一个羟基,得到部分羟基被保护的原人参三醇受体,反应路线如下所示:
式中,R1、R2和R3独立地选自取代或未取代的C6-10芳香烃类酰基、取代或未取代的C2-6烷酰基、硅基、取代或未取代的C1-10的烷基、取代或未取代的C3-10的环烷基、取代或未取代的C3-10的烯基、取代或未取代的C6-10的芳基,且R1、R2和R3中任一个为H;
所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
4.如权利要求1或3所述的制备方法,其特征在于,所述步骤(a)中,首先以保护基封闭原人参三醇中活性最高的12位羟基;然后用保护基封闭活性居中的3位羟基,得到6和20位羟基裸露的原人参三醇受体;或
所述步骤(a)中,首先在原人参三醇中的3,6,12位同时引入相同的保护基,然后选择性脱除12位上的保护基,得到12和20位羟基裸露的原人参三醇受体;或
所述步骤(a)中,首先以保护基封闭原人参三醇中活性最高的12位羟基;然后用保护基封闭活性居中的3位羟基,再用保护基保护活性最低的6位羟基,最后选择性脱除3位的保护基,得到3位和20位双羟基裸露的原人参三醇受体。
5.如权利要求1所述的方法,其特征在于,所述糖苷化反应是在惰性气体保护下,在有机溶剂中,在分子筛存在下,由糖基给体和步骤(a)得到的部分羟基被保护的原人参三醇受体,在路易斯酸促进下进行的反应。
6.如权利要求5所述的方法,其特征在于,所述惰性气体是氮气、氩气或氦气。
7.如权利要求5所述的方法,其特征在于,所述有机溶剂为甲苯、二氯甲烷、乙醚、或乙腈。
8.如权利要求5所述的方法,其特征在于,所述分子筛为 分子筛或酸洗的 分子筛。
9.如权利要求5所述的方法,其特征在于,所述为路易斯酸为含金的络合物,所述路易斯酸的用量为所述部分羟基被保护的原人参三醇受体用量的0.01-1当量。
10.如权利要求1所述的方法,其特征在于,所述方法还包括对步骤(c)制备得到的所述原人参三醇类人参皂苷进行分离纯化的步骤。
说明书
技术领域
本发明涉及6及12位单糖链和3/20以及6/20位双糖链-20(S)-原人参三醇类人参皂苷的合成。
背景技术
人参作为名贵中药在中国使用已达两千年之久,随着现代生物技术的发展,人参中所含人参皂苷已被确定为人参的主要活性成分(Sticher,O.ChemTech 1998,4,26.)。
到目前为止,从人参中分离鉴定的人参皂苷已达200多种,按照皂苷中所含苷元的结构不同,人参皂苷主要可以分为两类:原人参二醇类人参皂苷(Panaxadiols,PPDS)和原人参三醇类人参皂苷(Panaxatriols,PPTS)((a)Christensen,L.P.Adv.Food Nutri.Res.2009,55,1.(b)Qi,L.-W.;Wang,C.-Z.;Yuan,C.-S.Nat.Prod.Rep.2011,28,467.),结构如下:
尽管初步研究表明人参是通过其所含种类丰富的人参皂苷来发挥其药用活性 的,对一些纯的人参皂苷也已经进行了初步的生物活性测试,但由于极具药用前景的人参皂苷天然存在的极不均一性,使得单一人参皂苷的分离纯化非常困难。不仅如此,一些人参皂苷的天然含量还非常低,这进一步增加了单一人参皂苷的获取难度。正是由于高纯、单一人参皂苷从天然中获取的困难性,使得人参皂苷生物活性作用机理的研究难以进行。
为了克服人参皂苷样品的获取问题,人们已经发展了两种方法:
一是化学合成方法,这一方法主要用于合成难度较小的原人参二醇类人参皂苷的合成((a)Atopkina,L.N.;Denisenko,V.A.;Uvarova,N.I.;Elyakov,G.B.Carbohvdr.Res.1988,177,101.(b)Atopkina,L.N.;Uvarova,N.I.;Elyakov,G.B.Carbohydr.Res.1997,303,449.(c)Anufriev,V.P.;Malinovskaya,G.V.;Denisenko,V.A.;Uvarova,N.I.;Elyakov,G.B.;Kim,S.-II.;Beak,N.-I.Carbohydr.Res.1997,304,179.(d)Hui,Y.-Z.;Yang,Z.-Q.;Liu,J.-Y.;Teng,J.-J.;Xie,H.-Q.;Zhang,J.Chinese Patent,CN 1587273A.)。尽管合成难度较低,但由于缺乏有效的糖苷键构建方法,文献所报道的原人参二醇类人参皂苷的合成效率一般较低。
另一种是人参皂苷的酶法合成,这一方法主要是针对合成难度较大的原人参三醇类人参皂苷的合成((a)Danieli,B.;Falcone,L.;Monti,D.;Riva,S.;Gebhardt,S.;Schubert-Zsilavecz,M.J.Org.Chem.2001,66,262.(b)Ko,S.-R.;Choi,K.-J.;Suzuki,K.;Suzuki,Y.Chem.Pharm.Bull.2003,51,404.)。鉴于酶法合成中所涉及到酶一般价格昂贵且不易获取,因此酶法合成也不是理想解决样品供应的方法。
因此,尽管化学合成方法部分地解决了原人参二醇类人参皂苷的样品供应问题,但对于原人参三醇类人参皂苷的化学合成,在国内外鲜有文献报道。
与三醇类人参皂苷供应瓶颈形成鲜明对比的是,近来通过对三醇类人参皂苷活性研究发现,该类人参皂苷在抑制白血病细胞增殖(Popovich,D.G.;Kitts,D.D.Arch.Biochem.Biophys.2002,406,1.)、免疫调节(Yu,J.-L.;Dou,D.-Q.;Chen,X.-H.;Yang,H.-Z.;Guo,N.;Cheng,G.-F.Planta Med.2005,71,202)等方面表现出优异的药用前景,因此急需发展一种可靠、高效提供原人参三醇类人参皂苷的化学合成方法。
发明内容
本发明的目的在于提供一种原人参三醇类人参皂苷的化学合成方法。
本发明的第一方面,提供一种原人参三醇类人参皂苷的制备方法,包括以下步骤:
(a)以原人参三醇为原料,对原人参三醇的羟基进行保护,从而裸露20位羟基以及裸露3位、6位和12位中至少一个羟基,得到部分羟基被保护的原人参三醇受体;
(b)步骤(a)得到的部分羟基被保护的原人参三醇受体与全保护的糖基给体发生糖苷化反应,得到连接有全保护的糖基、且部分羟基被保护的原人参三醇类人参皂苷;
(c)对步骤(b)得到的所述连接有全保护的糖基、且部分羟基被保护的原人参三醇类人参皂苷,进行脱保护,从而得到原人参三醇类人参皂苷。
在另一优选例中,所述步骤(a)对原人参三醇的羟基进行保护为对原人参三醇的羟基进行区域选择性保护。
在另一优选例中,所述的糖基给体的结构如下所示:
式中,R5为C1-6的烷基、C3-10环烷基或C3-10芳基;
R6为全保护的β-D-葡萄糖基、α-D-葡萄糖基、β-D-半乳糖基、α-D-半乳糖基、β-D-甘露糖基、α-D-甘露糖基、β-D-木糖基、α-D-木糖基、β-D-2-氨基葡萄糖基、α-D-2-氨基葡萄糖基、α-L-鼠李糖基、β-L-鼠李糖基、α-D-阿拉伯糖基、β-D-阿拉伯糖基、α-L-阿拉伯糖基、β-L-阿拉伯糖基、α-L-岩藻糖基、β-L-岩藻糖基、β-D-葡萄糖醛酸基、α-D-葡萄糖醛酸基、β-D-半乳糖醛酸基、或α-D-半乳糖醛酸基;所述全保护是指用乙酰基、苯甲酰基、苄基、或硅基对全部的羟基进行保护。
在另一优选例中,所述硅基为三甲基硅烷基、三乙基硅烷基、叔丁基二甲硅烷基、或叔丁基二苯基硅基。
在另一优选例中,所述R5为正丁基、或环丙基。
在另一优选例中,所述步骤(a)中以原人参三醇为原料,对原人参三醇的羟基进行保护,从而裸露20位羟基、以及裸露3位、6位、20位中至少一个羟基,得到部分羟基被保护的原人参三醇受体,反应路线如下所示:
式中,R1、R2和R3独立地选自取代或未取代的C6-10芳香烃类酰基、取代或未取代的C2-6烷酰基、硅基、取代或未取代的C1-10的烷基、取代或未取代的C3-10的环烷基、取代或未取代的C3-10的烯基、取代或未取代的C6-10的芳基,且R1、R2和R3中任一个为H;
所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
所述卤素为F、Cl、Br或I。
在另一优选例中,所述芳香烃类酰基为苯甲酰基。
在另一优选例中,所述取代的芳香烃类酰基为对甲氧基苯甲酰基、对氯苯甲酰基、对溴苯甲酰基、对碘苯甲酰基、或对硝基苯甲酰基。
在另一优选例中,所述的硅基为三甲基硅烷基、三乙基硅烷基、叔丁基二甲硅烷基、或叔丁基二苯基硅基。
在另一优选例中,所述对羟基进行保护在有机溶剂中进行,所述有机溶剂为二氯甲烷、甲苯、四氢呋喃、或二甲基甲酰胺。
在另一优选例中,所述步骤(a)中,首先以保护基封闭原人参三醇中活性最高的12位羟基;然后用保护基封闭活性居中的3位羟基,得到6和20位羟基裸露的原人参三醇受体;或
所述步骤(a)中,首先在原人参三醇中的3,6,12位同时引入相同的保护基,然后选择性脱除12位上的保护基,得到12和20位羟基裸露的原人参三醇受体;或
所述步骤(a)中,首先以保护基封闭原人参三醇中活性最高的12位羟基;然后用保护基封闭活性居中的3位羟基,再用保护基保护活性最低的6位羟基,最后选择性脱除3位的保护基,得到3位和20位双羟基裸露的原人参三醇受体。
在另一优选例中,步骤(a)中的保护基选自:叔丁基二甲基硅基(TBS),乙酰基Ac、三甲基乙酰基、烯丙基。
在另一优选例中,步骤(c)中,采用四丁基氟化铵TBAF、三(二乙胺基)二氟三甲基锍硅酸盐TASF、HF吡啶络合物或对甲苯磺酸TSOH脱除硅基保护基。
在另一优选例中,步骤(c)中,脱除烯丙基保护基所用钯金属催化剂为Pd(OAc)2、PdCl2或Pd(PPh3)4。
在另一优选例中,步骤(c)中,在碱性条件下脱除酰基保护基,所述碱为NaOMe、氢氧化钠、氢氧化锂或氢氧化钾。
在另一优选例中,所有糖苷化反应是在惰性气体保护下,在有机溶剂中,在分子筛存在下,由糖基给体和步骤(a)得到的部分羟基被保护的原人参三醇受体,在路易斯酸促进下进行的反应。
在另一优选例中,所述惰性气体是氮气、氩气或氦气。
在另一优选例中,所述惰性气体是高纯的氮气、高纯的氩气或高纯的氦气。
在另一优选例中,所述高纯是指气体的纯度≥99.999%,其中,所述气体为氮气、氩气或氦气。
在另一优选例中,所述有机溶剂为甲苯、二氯甲烷、乙醚、或乙腈。
在另一优选例中,所述有机溶剂为干燥的甲苯、二氯甲烷、乙醚、或乙腈,所述干燥即指有机溶剂为无水有机溶剂或水含量≤500ppm的有机溶剂。
在另一优选例中,所述有机溶剂为二氯甲烷。
在另一优选例中,所述分子筛为 分子筛或酸洗的 分子筛。
在另一优选例中,所述分子筛为活化的 分子筛或酸洗的 分子筛。
在另一优选例中,所述为路易斯酸为含金的络合物,所述路易斯酸的用量为所述部分羟基被保护的原人参三醇受体用量的0.01-1当量。
在另一优选例中,所述步骤(b)中糖苷化反应时间为0.5-12小时。
在另一优选例中,所述含金的络合物为PPh3AuOTf或PPh3AuNTf2,较佳地,为PPh3AuNTf2,便于操作。
在另一优选例中,所述路易斯酸的用量为部分羟基被保护的原人参三醇受体用量的0.05-0.5当量,较佳地,为0.1-0.2当量。
在另一优选例中,所述方法还包括对步骤(c)制备得到的所述原人参三醇类人参皂苷进行分离纯化的步骤。
在另一优选例中,所述纯化包括柱层析纯化。
本发明的第二方面,提供一种原人参三醇类人参皂苷的制备方法,包括以下步骤:
(a)以原人参三醇为原料,对原人参三醇的羟基进行保护,从而裸露1~3个羟 基,得到部分羟基被保护的原人参三醇受体;
(b)步骤(a)得到的部分羟基被保护的原人参三醇受体与全保护的糖基给体发生糖苷化反应,得到连接有全保护的糖基、且部分羟基被保护的原人参三醇类人参皂苷;
(c)对步骤(b)得到的所述连接有全保护的糖基、且部分羟基被保护的原人参三醇类人参皂苷,进行脱保护,从而得到原人参三醇类人参皂苷。
在另一优选例中,步骤(a)中裸露20位羟基。
在另一优选例中,步骤(a)中裸露20位羟基以及裸露3位、6位和12位中至少一个羟基。
在另一优选例中,所述的糖基给体的结构如下所示:
式中,R5为C1-6的烷基、C3-10环烷基或C3-10芳基;
R6为全保护的β-D-葡萄糖基、α-D-葡萄糖基、β-D-半乳糖基、α-D-半乳糖基、β-D-甘露糖基、α-D-甘露糖基、β-D-木糖基、α-D-木糖基、β-D-2-氨基葡萄糖基、α-D-2-氨基葡萄糖基、α-L-鼠李糖基、β-L-鼠李糖基、α-D-阿拉伯糖基、β-D-阿拉伯糖基、α-L-阿拉伯糖基、β-L-阿拉伯糖基、α-L-岩藻糖基、β-L-岩藻糖基、β-D-葡萄糖醛酸基、α-D-葡萄糖醛酸基、β-D-半乳糖醛酸基、或α-D-半乳糖醛酸基;所述全保护是指用乙酰基、苯甲酰基Bz、苄基、或硅基对全部的羟基进行保护。
在另一优选例中,所述R5为正丁基、或环丙基。
本发明的第三方面,提供一种糖基给体在制备原人参三醇类人参皂苷中的应用,所述的糖基给体的结构如下所示:
式中,R5为C1-6的烷基、C3-10环烷基或C3-10芳基;
R6为全保护的β-D-葡萄糖基、α-D-葡萄糖基、β-D-半乳糖基、α-D-半乳糖基、β-D-甘露糖基、α-D-甘露糖基、β-D-木糖基、α-D-木糖基、β-D-2-氨基葡萄糖基、α-D-2-氨基葡萄糖基、α-L-鼠李糖基、β-L-鼠李糖基、α-D-阿拉伯糖基、β-D-阿拉伯糖基、α-L-阿拉伯糖基、β-L-阿拉伯糖基、α-L-岩藻糖基、β-L-岩藻糖基、β-D-葡萄糖醛酸基、α-D-葡萄糖醛酸基、β-D-半乳糖醛酸基、或α-D-半乳糖醛酸基;所述全保护是指用乙酰基、苯甲酰基Bz、苄基、或硅基对全部的羟基进行保护。
在另一优选例中,所述R5为正丁基、或环丙基。
本发明提供了一种新颖的化学合成原人参三醇类人参皂苷的方法,方法可靠、高效、收率及立体选择性高。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一赘述。
具体实施方式
本申请的发明人经过广泛而深入地研究,首次以原人参三醇为原料,化学合成了6位连有单糖链、12位连有单糖链、6位和20位连有双糖链、或3位和20位连有双糖链的人参皂苷。尽管12位羟基最为活泼,但是3位、6位、12位羟基的活性差别不大,很难对羟基进行区域选择性保护,因此难以在3位、6位、12位羟基处实现糖苷化,本发明人意外发现,通过选用特殊的取代基可以对羟基进行区域选择性保护,并进一步选用糖基炔酯给体引入糖链,可以得到结构稳定的原人参三醇类人参皂苷,本发明的方法新颖、可靠,高效,得到的人参皂苷的收率及立体选择性高。在此基础上,完成了本发明。
人参皂苷(又称人参皂甙)
人参皂甙是人参的主要成分。目前,通过分离得到的人参皂甙,命名为Ro、Ra1、Ra2、Rb1、Rb2、Rb3、Rc、Rd、Rf、Rg1、Rg2、Rg3、Rh1、Rh2等。到目前为止从人参中分离鉴定的人参皂苷的数量已超过200种,皆由甙元和糖链两部分组成。其中除人参皂苷Ro的甙元是齐墩果酸外,其它人参皂苷的甙元主要是原人参二醇和原人参三醇。人参的神秘功效正是通过其中的人参皂苷来实现的。 由于人参皂苷天然存在的高度不均一性,使得单一人参皂苷的获取十分困难。因此,对于人参皂苷的活性研究也主要限制在几种天然含量较高且便于分离的人参皂苷之中。从而大大制约了人参这一名贵中药的进一步开发利用。因此,通过化学手段实现人参皂苷的高效合成无疑是解决制约人参皂苷的药用研究的最为有效的手段。
人参皂苷的制备方法
本发明提供的原人参三醇类人参皂苷的制备方法,包括以下步骤:
(a)以原人参三醇为原料,对原人参三醇的羟基进行保护,从而裸露20位羟基以及裸露3位、6位和12位中至少一个羟基,得到部分羟基被保护的原人参三醇受体;
(b)步骤(a)得到的部分羟基被保护的原人参三醇受体与全保护的糖基给体发生糖苷化反应,得到连接有全保护的糖基、且部分羟基被保护的原人参三醇类人参皂苷;
(c)对步骤(b)得到的所述连接有全保护的糖基、且部分羟基被保护的原人参三醇类人参皂苷,进行脱保护,从而得到原人参三醇类人参皂苷。
在另一优选例中,所述步骤(a)对原人参三醇的羟基进行保护为对原人参三醇的羟基进行区域选择性保护。
在另一优选例中,所述的糖基给体的结构如下所示:
式中,R5为C1-6的烷基、C3-10环烷基或C3-10芳基;
R6为全保护的β-D-葡萄糖基、α-D-葡萄糖基、β-D-半乳糖基、α-D-半乳糖基、β-D-甘露糖基、α-D-甘露糖基、β-D-木糖基、α-D-木糖基、β-D-2-氨基葡萄糖基、α-D-2-氨基葡萄糖基、α-L-鼠李糖基、β-L-鼠李糖基、α-D-阿拉伯糖基、β-D-阿拉伯糖基、α-L-阿拉伯糖基、β-L-阿拉伯糖基、α-L-岩藻糖基、β-L-岩藻糖基、β-D-葡萄糖醛酸基、α-D-葡萄糖醛酸基、β-D-半乳糖醛酸基、或α-D-半乳糖醛酸基;所述全保护是指用乙酰基、苯甲酰基、苄基、或硅基对全部的羟基进行保护。
在另一优选例中,所述R5为正丁基、或环丙基。
在另一优选例中,所述步骤(a)中以原人参三醇为原料,对原人参三醇的羟基进行保护,从而裸露20位羟基、以及裸露3位、6位、20位中至少一个羟基,得到部分羟基被保护的原人参三醇受体,反应路线如下所示:
式中,R1、R2和R3独立地选自取代或未取代的C6-10芳香烃类酰基、取代或未取代的C2-6烷酰基、硅基、取代或未取代的C1-10的烷基、取代或未取代的C3-10的环烷基、取代或未取代的C3-10的烯基、取代或未取代的C6-10的芳基,且R1、R2和R3中任一个为H;
所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
所述卤素为F、Cl、Br或I。
在另一优选例中,所述芳香烃类酰基为苯甲酰基。
在另一优选例中,所述取代的芳香烃类酰基为对甲氧基苯甲酰基、对氯苯甲酰基、对溴苯甲酰基、对碘苯甲酰基、或对硝基苯甲酰基。
在另一优选例中,所述的硅基为三甲基硅烷基、三乙基硅烷基、叔丁基二甲硅烷基、或叔丁基二苯基硅基。
在另一优选例中,所述对羟基进行保护在有机溶剂中进行,所述有机溶剂为二氯甲烷、甲苯、四氢呋喃、或二甲基甲酰胺。
在另一优选例中,所述步骤(a)中,首先以保护基封闭原人参三醇中活性最高的12位羟基;然后用保护基封闭活性居中的3位羟基,得到6和20位羟基裸露的原人参三醇受体;或
所述步骤(a)中,首先在原人参三醇中的3,6,12位同时引入相同的保护基,然后选择性脱除12位上的保护基,得到12和20位羟基裸露的原人参三醇受体;或
所述步骤(a)中,首先以保护基封闭原人参三醇中活性最高的12位羟基;然后用保护基封闭活性居中的3位羟基,再用保护基保护活性最低的6位羟基,最后选择性脱除3位的保护基,得到3位和20位双羟基裸露的原人参三醇受体。
在另一优选例中,步骤(a)中的保护基选自:叔丁基二甲基硅基(TBS),乙酰 基Ac、三甲基乙酰基、烯丙基。
在另一优选例中,步骤(c)中,采用四丁基氟化铵TBAF、三(二乙胺基)二氟三甲基锍硅酸盐TASF、HF吡啶络合物或对甲苯磺酸TSOH脱除硅基保护基。
在另一优选例中,步骤(c)中,脱除烯丙基保护基所用钯金属催化剂为Pd(OAc)2、PdCl2或Pd(PPh3)4。
在另一优选例中,步骤(c)中,在碱性条件下脱除酰基保护基,所述碱为NaOMe、氢氧化钠、氢氧化锂或氢氧化钾。
在另一优选例中,所述糖苷化反应是在惰性气体保护下,在有机溶剂中,在分子筛存在下,由糖基给体和步骤(a)得到的部分羟基被保护的原人参三醇受体,在路易斯酸促进下进行的反应。
所述惰性气体是氮气、氩气或氦气。
在另一优选例中,所述惰性气体是高纯的氮气、高纯的氩气或高纯的氦气。
在另一优选例中,所述高纯是指气体的纯度≥99.999%,其中,所述气体为氮气、氩气或氦气。
在另一优选例中,所述有机溶剂为甲苯、二氯甲烷、乙醚、或乙腈。
在另一优选例中,所述有机溶剂为干燥的甲苯、二氯甲烷、乙醚、或乙腈,所述干燥即指有机溶剂为无水有机溶剂或水含量≤500ppm的有机溶剂。
在另一优选例中,所述有机溶剂为二氯甲烷。
在另一优选例中,所述分子筛为 分子筛或酸洗的 分子筛。
在另一优选例中,所述分子筛为活化的 分子筛或酸洗的 分子筛。
在另一优选例中,所述为路易斯酸为含金的络合物,所述路易斯酸的用量为所述部分羟基被保护的原人参三醇受体用量的0.01-1当量。
在另一优选例中,所述步骤(b)中糖苷化反应时间为0.5-12小时。
在另一优选例中,所述含金的络合物为PPh3AuOTf或PPh3AuNTf2,较佳地,为PPh3AuNTf2,便于操作。
在另一优选例中,所述路易斯酸的用量为部分羟基被保护的原人参三醇受体用量的0.05-0.5当量,较佳地,为0.1-0.2当量。
在另一优选例中,所述方法还包括对步骤(c)制备得到的所述原人参三醇类人参皂苷进行分离纯化的步骤。
在另一优选例中,所述纯化包括柱层析纯化。
在另一优选例中,本发明的人参皂苷的制备方法,包括步骤:
(a)以原人参三醇为原料,对3位和12位、或3位和6位、或6位和12位的羟基进行保护,裸露6位和20位、或12位和20位、或3位和20位羟基,得到部分羟基被保护的原人参三醇受体;
(b)步骤(a)得到的所述部分羟基被保护的原人参三醇受体,在其6位、12位、6位和20位、或3位和20位的羟基处与糖基给体发生糖苷化反应,得到在6位、12位、6位和20位、或3位和20位与全保护的糖基连接的被保护的人参皂苷;
(c)脱除步骤(b)得到的所述与全保护的糖基连接的被保护的人参皂苷上的全部保护基团,得到6位连有单糖链、12位连有单糖链、6位和20位连有双糖链、或3位和20位连有双糖链的人参皂苷。
在另一优选例中,本发明的人参皂苷的制备方法,包括步骤:
(1)以原人参三醇为原料,裸露6位和20位、或12位和20位、或3位和20位羟基,对3位和12位、或3位和6位、或6位和12位的羟基进行保护,得到部分羟基被保护的原人参三醇受体式A1化合物、式A2化合物、式A3化合物、或A4化合物,结构如下所示:
(2)式A1化合物、式A2化合物、式A3化合物、或式A4化合物与糖基受体发生糖苷化反应,在式A1化合物的6位、在式A2化合物的12位、在式A3化合物的3位和20位、或在式A4化合物6位和20位,引入全保护的糖基,得到式I1化合物、式I2化合物、式I3化合物、或式I4化合物,其结构如下:
(3)脱除式I1化合物、式I2化合物、式I3化合物、或式I4化合物所有保 护基,得到6位连有单糖链的人参皂苷式II1化合物、12位连有单糖链的人参皂苷式II2化合物、6位和20位连有双糖链的人参皂苷式II3化合物、或3位和20位连有双糖链的人参皂苷式II4化合物,结构如下:
上式中,R1、R2独立地选自取代或未取代的C6-10芳香烃类酰基、取代或未取代的C2-6烷酰基、硅基、取代或未取代的C1-10的烷基、取代或未取代的C6-10的芳基、取代或未取代的C3-10的环烷基、取代或未取代的C3-10的烯基;所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
R31为大位阻硅基、取代或未取代的C6-10芳香烃类酰基、或取代或未取代的C2-6烷酰基;所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
R32为小位阻硅基、取代或未取代的C1-10的烷基、取代或未取代的C3-10的环烷基、取代或未取代的C3-10的烯基、取代或未取代的C6-10的芳基;所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
R为羟基全部被保护的β-D-葡萄糖基、α-D-葡萄糖基、β-D-半乳糖基、α-D-半乳糖基、β-D-甘露糖基、α-D-甘露糖基、β-D-木糖基、α-D-木糖基、β-D-2-氨基葡萄糖基、α-D-2-氨基葡萄糖基、α-L-鼠李糖基、β-L-鼠李糖基、α-D-阿拉伯糖基、β-D-阿拉伯糖基、α-L-阿拉伯糖基、β-L-阿拉伯糖基、α-L-岩藻糖基、β-L-岩藻糖基、β-D-葡萄糖醛酸基、α-D-葡萄糖醛酸基、β-D-半乳糖醛酸基、或α-D-半乳糖醛酸基;所述羟基全部被保护是指用乙酰基、苯甲酰基、苄基、或硅基对全部的羟基进行保护。
R’为羟基全部裸露的β-D-葡萄糖基、α-D-葡萄糖基、β-D-半乳糖基、α-D-半乳糖基、β-D-甘露糖基、α-D-甘露糖基、β-D-木糖基、α-D-木糖基、β-D-2-氨基葡萄糖基、α-D-2-氨基葡萄糖基、α-L-鼠李糖基、β-L-鼠李糖基、α-D-阿拉伯糖基、β-D-阿拉伯糖基、α-L-阿拉伯糖基、β-L-阿拉伯糖基、α-L-岩藻糖基、β-L-岩藻糖基、β-D- 葡萄糖醛酸基、α-D-葡萄糖醛酸基、β-D-半乳糖醛酸基、或α-D-半乳糖醛酸基。
在另一优选例中,R1、R2独立地选自苯甲酰基、对甲氧基苯甲酰基、对硝基苯甲酰基、硅基、苄基、苯基、C3-6的烯基、C1-6的烷酰基、C1-6的烷基。
所述大位阻硅基优选为三乙基硅烷基、叔丁基二甲硅烷基、或叔丁基二苯基硅基。
所述小位阻硅基优选为三甲基硅基。
在另一优选例中,R31为C7-10芳基酰基、C1-6的烷酰基、二甲基叔丁基硅基、三乙基硅基、或二苯基叔丁基硅基。
在另一优选例中,R31为苯甲酰基、对甲氧基苯甲酰基、对硝基苯甲酰基、或三甲基乙酰基。
在另一优选例中,R32为三甲基硅基、苄基、苯基、C3-6的烯基、或C1-6的烷基。
在另一优选例中,R32为烯丙基。
在另一优选例中,原人参三醇受体式A1化合物的制备,包括步骤
(i)在有机溶剂中,原人参三醇和酰卤在碱的作用下反应,引入12位羟基的保护基R31,得到12位羟基被保护的原人参三醇;
(ii)在有机溶剂中,所述12位羟基被保护的原人参三醇与酰卤、硅基氯、或卤代烃在碱的作用下反应,引入3位羟基的保护基R1,得到式A1化合物。
R31、R1的定义如前所述。
在另一优选例中,R31为苯甲酰基、对甲氧基苯甲酰基、对硝基苯甲酰基、或三甲基乙酰基。
在另一优选例中,R31为三甲基乙酰基。
在另一优选例中,R1为叔丁基二甲基硅基。
所述有机溶剂为C1-4的卤代烷,较佳地,为C1-4的氯代烷,更佳地,为二氯甲烷。
所述的酰卤为C7-10芳基酰卤、取代的芳基酰卤、C2-10烷基酰卤;所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
在另一优选例中,所述酰卤为苯甲酰氯、对甲基苯甲酰氯、对硝基苯甲酰氯、氯乙酰、三甲基乙酰氯。
所述的硅基氯为C3-16烷基取代的硅基氯或C6-20芳基取代的硅基氯。
所述的卤代烃为C1-10的卤代烷、C2-10的卤代烯烃、C3-10的卤代炔烃、C6-10的卤代芳烃,所述卤代为氯代、溴代或碘代。
所述的碱为吡啶、三乙胺、DMAP、咪唑、氢化钠、氢化钾、LDA或LiHMDS。
由酰卤引入保护基R31或R1,所用的碱优选为吡啶、三乙胺、DMAP。
由硅基氯引入保护基R1,所用的碱优选为三乙胺、或咪唑。
由卤代烃引入保护基R1,所用的碱优选为氢化钠、氢化钾、LDA或LiHMDS。
原料中原人参三醇和含保护基的试剂(如酰卤)的摩尔比为1∶0.9-10,较佳地,为1∶0.9-5。更佳地,为1∶1-1.5。反应时间为0.5-24小时,较佳地,为1-12小时。反应温度为-20~50℃,较佳地,为-10~30℃,更佳地,为0~25℃。
12位羟基被保护的原人参三醇和含保护基的试剂(如酰卤、硅基氯或卤代烃)的摩尔比为1∶0.9-10,较佳地,为1∶0.9-5。更佳地,为1∶1-1.5。反应时间为0.5-24小时,较佳地,为1-12小时。反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃。
在另一优选例中,原人参三醇受体式A2化合物的制备,包括步骤:
(a)原人参三醇和含保护基的试剂在碱的作用下反应,一次引入3、6和12位羟基上的保护基R1、R2、R30;
(b)选择性脱除活性最高12位羟基上的保护基R30得到原人参三醇受体式A2化合物。
上述步骤(a)中,R1、R2的定义如前所述,且R1=R2=R30。
所述含保护基的试剂为酸酐、酰卤、硅基氯或卤代烃;
所述的碱为吡啶、三乙胺、DMAP、咪唑、氢化钠、氢化钾、LDA或LiHMDS;
由酰卤引入保护基R1、R2、R30,所用的碱优选为吡啶、三乙胺、DMAP。
由硅基氯引入保护基R1、R2、R30,所用的碱优选为三乙胺、或咪唑。
由卤代烃引入保护基R1、R2、R30,所用的碱优选为氢化钠、氢化钾、LDA或LiHMDS。
原人参三醇和含保护基的试剂(如酸酐、酰卤、硅基氯、或卤代烃)的摩尔比为1∶3.0-40,较佳地,为1∶3.0-30,更佳地,为1∶4.0-20;
反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃。
在另一优选例中,所述的酰卤为C7-10芳基酰卤、取代的芳基酰卤、C2-10烷基 酰卤;所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
在另一优选例中,所述酰卤为苯甲酰氯、对甲基苯甲酰氯、对硝基苯甲酰氯、氯乙酰、或三甲基乙酰氯。
在另一优选例中,所述的酸酐为乙酸酐Ac2O。
在另一优选例中,所述R1、R2、R30为乙酰基。
上述步骤(b)中,选择性脱除12羟基上的R30,其中,
R30为酰基时,在碱性条件下脱除R30酰基保护基,所述碱为NaOMe、氢氧化钠、氢氧化锂或氢氧化钾;较佳地,为甲醇钠(NaOMe);
R30为硅基时,采用四丁基氟化铵TBAF、三(二乙胺基)二氟三甲基锍硅酸盐TASF、HF吡啶络合物或对甲苯磺酸TSOH脱除硅基保护基;
R30为烃基时,如烯丙基保护基,采用钯金属催化剂Pd(OAc)2、PdCl2或Pd(PPh3)4脱除R30。
反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃;
反应时间为0.5-24小时,较佳地,为1-12小时。
在另一优选例中,原人参三醇受体式A2化合物的制备,包括步骤:
(a)原人参三醇和卤代烃在碱的作用下选择性引入12位羟基上的保护基R30;
(b)在步骤a的产物上一次性引入3、6位羟基上的保护基R1及R2(R1=R2);
(c)然后选择性脱除12羟基上保护基R30得到原人参三醇受体式A2化合物。
R1、R2选自:取代或未取代的C6-10芳香烃类酰基、取代或未取代的C2-6烷酰基、硅基、取代或未取代的C1-10的烷基、取代或未取代的C6-10的芳基、取代或未取代的C3-10的环烷基、取代或未取代的C3-10的烯基;所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
R30为C1-10烃基,优选为苄基、苯基、C3-6的烯基、或C1-6的烷基。
在另一优选例中,步骤(a)所述的碱为吡啶、三乙胺、DMAP、咪唑、氢化钠、氢化钾、LDA或LiHMDS;优选为氢化钠、氢化钾、LDA或LiHMDS。
原人参三醇和卤代烃的摩尔比为1∶1.0-10,较佳地,为1∶1.0-5。更佳地,为 1∶1-1.5。反应时间为0.5-24小时,较佳地,为1-12小时。反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃。
步骤a的产物和含保护基的试剂(如酰卤、硅基氯、或卤代烃)的摩尔比为1∶2.0-40,较佳地,为1∶2.0-30,更佳地,为1∶3.0-20;
反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃。
在另一优选例中,原人参三醇受体式A3化合物的制备,包括步骤:
(a)在有机溶剂中,原人参三醇和卤代烃在碱的作用下反应,在原人参三醇12位羟基处引入保护基R32;
(b)在有机溶剂中,步骤(a)的产物与硅基氯在碱的作用下反应,在3位羟基处引入硅基保护基;
(c)步骤(b)的产物与酸酐、酰卤、硅基氯、或卤代烃在碱的作用下反应,引入6位羟基的保护基R2;
(d)脱除步骤(c)的产物的3位上的硅基保护基得到式A3化合物。
R32为C1-10的烷基、C2-10的烯基、C3-10的炔基、C6-10的芳基。
R2的定义如前所述。
在另一优选例中,R32为烯丙基。
在另一优选例中,所述步骤(b)在3位羟基处引入硅基保护基中,所述硅基保护基为三甲基硅烷基、三乙基硅烷基、叔丁基二甲硅烷基、或叔丁基二苯基硅基,优选为叔丁基二甲硅烷基。
在另一优选例中,R2为乙酰基。
步骤(a)中,所述卤代烃为C1-10的卤代烷、C2-10的卤代烯烃、C3-10的卤代炔烃、C6-10的卤代芳烃。
所述碱为氢化钠、氢化钾、LDA(二异丙基氨基锂)或LiHMDS(六甲基二硅基氨基锂);
所述原人参三醇三醇和卤代烃摩尔比为1∶0.9-10,较佳地,为1∶0.9-5。更佳地,为1∶1-1.5。反应时间为0.5-24小时,较佳地,为1-12小时。反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃。
步骤(b)中,所述的硅基氯为C3-16烷基取代的或C6-20芳基取代的硅基氯;
所用碱为三乙胺、咪唑;
步骤(a)所得产物和卤代烃摩尔比为1∶0.8-10,较佳地,为1∶1.0-5。更佳地,为1∶1-1.5。反应时间为0.5-24小时,较佳地,为1-12小时。反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃。
步骤(c)中,所述的酰卤为C7-10芳基酰卤、取代的芳基酰卤、C2-10烷基酰卤;所述的取代是指采用选自下组的基团进行单取代、二取代或三取代:C1-10的烷基、C1-10的烷氧基、C3-10的烯基、C3-10环烷基、C6-10芳基、硝基、卤素、氨基、硅基。
在另一优选例中,所述酰卤为苯甲酰氯、对甲基苯甲酰氯、对硝基苯甲酰氯、氯乙酰、三甲基乙酰氯。
所述酸酐为乙酸酐。
所述的硅基氯为C3-16烷基取代的或C6-20芳基取代的硅基氯。
所述卤代烃为C1-10的卤代烷、C2-10的卤代烯烃、C3-10的卤代炔烃、C6-10的卤代芳烃;
所用碱为吡啶、三乙胺及DMAP。
步骤(b)所得产物和卤代烃摩尔比为1∶0.1-10,较佳地,为1∶0.5-5。更佳地,为1∶1.5-4。反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃。
以上反应所用有机溶剂为C1-C4的卤代烷、或吡啶。
在另一优选例中,原人参三醇受体式A4化合物的制备,包括步骤:
(a)在原人参三醇活性最高的12位羟基上引入保护基R32,
(b)再引入三醇3位羟基上引入保护基R1即式A4化合物。
其中对R1、R32的定义如前所述。
在另一优选例中,R32为C1-10的烷基、C2-10的烯基、C3-10的炔基、C6-10的芳基。
在另一优选例中,R32为烯丙基。
在另一优选例中,R1为硅基,所述硅基选自三甲基硅烷基、三乙基硅烷基、叔丁基二甲硅烷基、或叔丁基二苯基硅基,优选为叔丁基二甲硅烷基。
步骤(a)中,原人参三醇和卤代烃在碱的作用下反应,在原人参三醇12位羟基处引入保护基R32。
所述卤代烃为C1-10的卤代烷、C2-10的卤代烯烃、C3-10的卤代炔烃、C6-10的卤代芳烃。
所述碱为氢化钠、氢化钾、LDA(二异丙基氨基锂)或LiHMDS(六甲基二硅基氨基锂);
所述原人参三醇三醇和卤代烃摩尔比为1∶0.1-3,较佳地,为1∶0.5-2。更佳地,为1∶1-1.5。反应时间为0.5-24小时,较佳地,为1-12小时。反应温度为-20~50℃,较佳地,为-10~25℃,更佳地,为0~20℃。
本发明的优点在于:
(1)提供一种新型、高效制备极具合成难度的6位连接糖基、12位连接糖基、3/20位连接糖基、6/20位连接糖基的-20(S)-原人参三醇类人参皂苷的化学方法。
(2)本发明不仅能够实现含有多个羟基的原人参三醇中多个羟基的高效区分,而且在构建糖苷键时收率及立体选择性都很高。
(3)本发明将大大推进原人参三醇类人参皂甙类化合物活性机理的研究及其药物开发的进程。
本发明提到的上述特征,或实施例提到的特征可以任意组合。本案说明书所揭示的所有特征可与任何组合物形式并用,说明书中所揭示的各个特征,可以被任何被提供相同、均等或相似目的的替代性特征取代。因此除有特别说明,所揭示的特征仅为均等或相似特征的一般性例子。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。本发明中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。如对羟基进行保护引入保护基的实验条件和脱除保护基的实验条件可以采用本领域惯常使用的羟基保护、脱保护条件。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
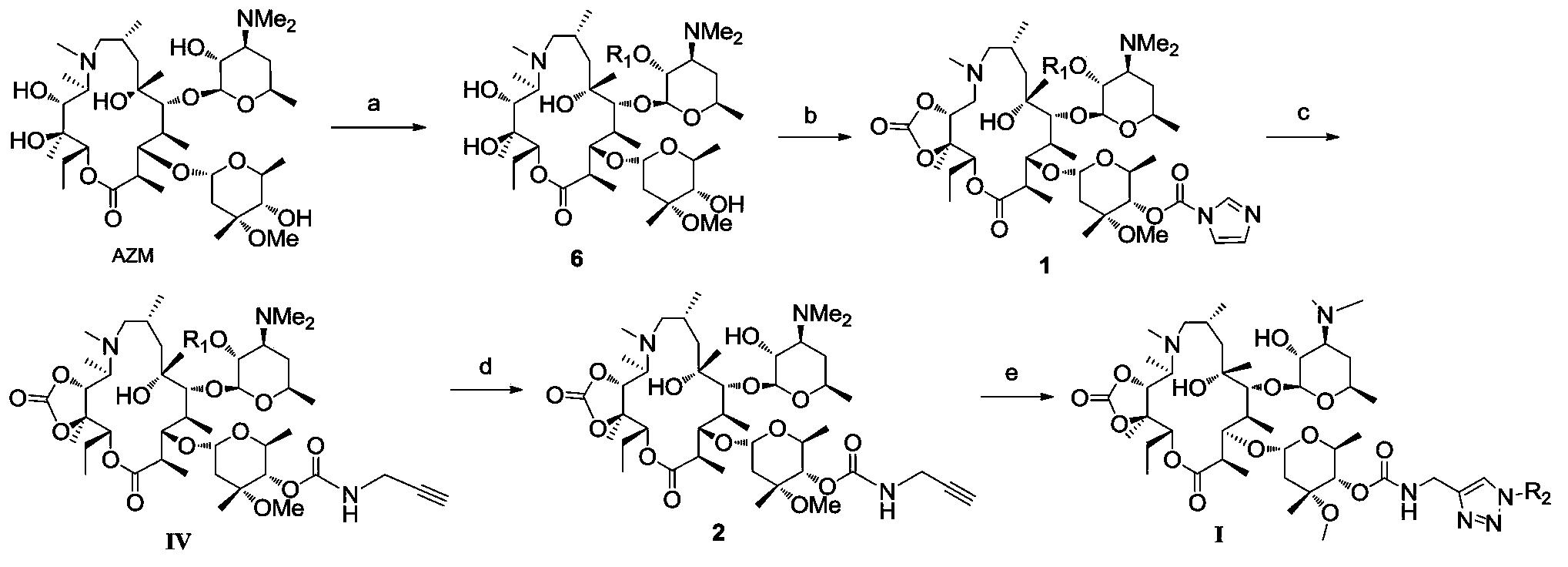
实施例1
人参皂甙Rh1的合成
合成路线如下所示:
试剂和条件:
a)三甲基乙酰氯PivCl,三乙基胺Et3N,二氯甲烷DCM,0℃,80%.
b)叔丁基二甲基氯硅烷TBSCl,咪唑imidazole,二甲基甲酰胺DMF,68%.
c)化合物4,PPh3AuNTf2(三苯基膦二(三氟甲磺酰胺基)金(I))(0.5当量equiv.), 分子筛MS,二氯甲烷DCM,室温rt,78%.
d)CSA(樟脑磺酸),CH2Cl2/MeOH,rt,91%.
e)10%KOH/MeOH,80%.
具体实验过程和数据:
氩气保护下,将原人参三醇(100mg,0.21mmol)溶于3ml无水DMF中,冰浴下缓慢滴加三乙胺(146μL,0.42mmol)和PivCl(52μL,0.42mmol),2小时候用水淬灭反应,加入大量乙酸乙酯萃取,水洗(3*30mL),饱和食盐水洗涤,无水硫酸钠干燥,后旋干可得粗产物,经柱层析纯化得到化合物2(收率80%)。
[α]25D=11.2(c 4.5,CHCl3);1H NMR(300MHz,CDCl3)δ5.06(m,1H),4.76-4.70(m,1H),4.05-4.00(m,1H),3.12(dd,J=4.5,8.1Hz,1H),2.14-1.81(m,8H),1.64(s,3H),1.55(s,3H),1.25(s,3H),1.14(s,9H),1.05(s,3H),1.03(s,3H),0.92(s,3H),0.91(s,3H),0.83(s,3H);13C NMR(75MHz,CDCl3)δ177.5,131.1,124.9,78.2,76.4,73.2,68.2,60.8,53.3,52.5,49.0,46.5,43.8,40.6,39.1,39.0,38.9,38.4,35.7,30.8,30.7,27.4,27.0,26.7,25.8,25.7,22.1,17.6,17.1(2C),17.0,15.4; HRMS(MALDI)计算C35H60O5Na[M+Na]+583.4330,实测583.4346.
氩气保护下,将化合物2(300mg,0.54mmol)、TBSCl(162mg,1.08mmol)和咪唑(74mg,1.08mmol)溶于2mL无水DMF,室温搅拌24h,大量乙酸乙酯稀释,水洗,饱和食盐水洗涤,无水硫酸钠干燥,过滤浓缩后柱层析(石油醚∶乙酸乙酯=10∶1)得化合物3(245mg,收率68%)。
[α]25D=18.2(c 0.85,CHCl3);1H NMR(300MHz,CDCl3)δ5.10(m,1H),4.77(m,1H),4.07(m,1H),3.15(dd,J=5.4,9.9Hz,1H),2.18-1.84(m,7H),1.68(s,3H),1.59(s,3H),1.19(s,3H),1.17(s,9H),1.09(s,3H),1.06(s,3H),0.95(s,3H),0.91(s,3H),0.89(s,3H),0.86(s,9H),0.00(s,6H);13C NMR(75MHz,CDCl3)δ177.4,131.1,125.0,79.0,76.5,73.2,68.5,61.1,53.4,52.6,49.2,46.6,43.9,40.6,39.8,39.1,38.8,38.5,35.7,30.9,27.5,27.2,27.1,26.8,25.9,25.7,25.6,22.2,18.0,17.6,17.2,17.1(2C),15.9,-3.7,-5.0;HRMS(MALDI)计算C41H74O5SiNa[M+Na]+697.5198,实测697.5220.
氩气保护下,将化合物3(20mg,0.030mmol)、化合物4(68mg,0.089mmol)和4A MS(88mg)溶于无水二氯甲烷中,室温搅拌半小时后加入催化剂PPh3AuNTf2(11mg,0.015mmol),室温搅拌半个小时后,停止反应,硅藻土过滤掉分子筛,减压浓缩后柱层析(甲苯∶乙酸乙酯=40∶1)得化合物5(29mg,78%)。
[α]25D=14.8(c 0.7,CHCl3);1H NMR(400MHz,CDCl3)δ8.03(d,J=5.4Hz,2H),7.92(t,J=6.0Hz,4H),7.80(d,J=5.4Hz,2H),7.56-7.23(m,12H),5.94(t,J=7.2Hz,1H),5.67(t,J=7.2Hz,1H),5.62(t,J=6.9Hz,1H),5.17(m,2H),4.75-4.70(m,1H),4.63(dd,J=1.8,9.3Hz,1H),4.53(dd,J=2.1,9.0Hz,1H),4.25(m,1H),4.05(dd,J=6.0,7.5Hz,1H),2.99(dd,J=3.9,8.1Hz,1H),2.22(bs,1H),2.05-1.93(m,4H),1.75(s,3H),1.65(s,3H),1.18(s,9H),1.09(s,3H),0.95(s,3H),0.90(s,3H),0.88(s,3H),0.83(s,3H),0.70(s,9H),0.65(s,3H),0.10(s,3H),0.21(s,3H);13C NMR(75MHz,CDCl3)δ177.3,166.0,165.8,165.2,165.1,133.4,133.2,133.0,131.1,129.8,129.7(2C),129.5,129.2,128.7,128.6,128.4,128.3,128.2,128.1,125.1,102.6,80.5,79.3,73.3,73.1,72.1,69.5,59.9,53.4,52.5,49.0,43.8,40.6,39.2,39.1,30.2,27.1,25.8(2C),22.2,17.9,17.6,17.2,17.1,16.8,15.9,-3.8,-5.3;HRMS(MALDI)计算C75H100O14SiNa[M+Na]+1275.6775,实测1275.6796.
将化合物5(70mg,0.056mmol)和CSA(23mg,0.099mmol)溶入10mL甲醇中,室温搅拌10h后停止反应,减压浓缩,柱层析(石油醚∶乙酸乙酯=3∶1) 得化合物6(57mg,91%)。
配制10%KOH/MeOH溶液(0.2g KOH,2.5mL MeOH),冷却至室温后,将上述溶液加入到化合物6(60mg,0.053mmol)中,室温搅拌12h后,酸性树脂淬灭反应,调节PH为7~8,过滤掉酸性树脂,减压浓缩后柱层析(二氯甲烷∶甲醇=10∶1)得化合物7Ginsenoside Rh1(27mg,80%)。
[α]25D=23.6(c 0.8,MeOH);1H NMR(300MHz,氘代吡啶pyridine-d5)δ5.34(m,1H),5.08(d,J=7.8Hz,1H),4.59(d,J=10.8Hz,1H),4.48-4.38(m,2H),4.33-4.23(m,2H),4.16(t,J=7.8Hz,1H),3.98-3.90(m,2H),3.58(dd,J=4.8,11.1Hz,1H),2.57(m,2H),2.32(m,2H),2.11(s,3H),1.67(s,3H),1.64(s,6H),1.41(s,3H),1.19(s,3H),1.04(s,3H),0.82(s,3H);13C NMR(100MHz,pyridine-d5)δ130.8,126.3,106.1,80.1,79.7,78.6,78.2,75.5,73.0,71.9,71.1,63.1,61.5,54.8,51.7,50.2,48.3,45.3,41.1,40.4,39.7,39.4,35.8,32.1,31.8,31.3,28.0,27.1,26.9,25.8,23.0,17.7(2C),17.4,16.8,16.4;HRMS(MALDI)计算C36H62O9Na[M+Na]+661.4286,实测661.4300.
实施例2
人参皂甙Chikusetsusaponin L10的合成
试剂和条件:
a)乙酸酐Ac2O,pyridine(吡啶),rt,87%;
b)NaOMe,MeOH,rt,30min,98%.
c)化合物4,PPh3AuNTf2(三苯基膦二(三氟甲磺酰胺基)金(I))(0.5equiv.), MS,DCM,rt,89%.
d)KOH,MeOH.
具体实验过程和数据:
氩气保护下,将化合物1(500mg,1.05mmol)溶于3mL无水吡啶,冰浴下缓慢滴加3mL乙酸酐,半小时后用1mol/L盐酸淬灭反应,大量乙酸乙酯稀释,饱和碳酸氢钠洗涤,水洗,饱和食盐水洗涤,无水硫酸钠干燥,过滤浓缩后柱层析(石油醚∶乙酸乙酯=6∶1)得化合物8(549mg,84%)。
[α]25D=22.3(c 1.0,CHCl3);1H NMR(300MHz,CDCl3)δ5.38-5.30(m,1H),5.16(m,1H),4.74(m,1H),4.50(dd,J=4.8,11.4Hz,1H),2.04(s,9H),1.71(s,3H),1.64(s,3H),1.15(s,3H),1.13(s,3H),1.02(s,3H),1.00(s,3H),0.97(s,3H),0.81(s,3H);13C NMR(100MHz,CDCl3)δ170.7,170.0,169.5,131.2,125.0,79.9,75.9,73.5,70.1,58.5,52.8,52.3,49.1,42.0,44.3,40.4,39.1,37.9,37.6,35.9,31.2,30.1,27.9,26.9,25.6,23.0,22.1,21.8,21.3,21.1,17.5,17.0(2C),16.7,16.6;HRMS(MALDI)计算C36H58O7Na[M+Na]+625.4096,实测625.4075.
氩气保护下,将NaOMe(13mg,0.249mmol)加入到3mL无水甲醇中,然后把化合物8(100mg,0.166mmol)加入到甲醇钠溶液中,室温搅拌45min后,停止反应,酸性树脂淬灭反应,调节PH为7~8,过滤掉酸性树脂,减压浓缩后柱层析(石油醚∶乙酸乙酯=2∶1)得化合物9(91mg,
一种原人参三醇类人参皂苷的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。





















动态评分
0.0