

IPC分类号 : C07C247/10,C07C247/12,C07C255/67,C07D333/58,C07D215/14,C07C247/14,C07C271/22,C07J1/00,C07J63/00,C07D307/00,C07D493/18









专利摘要
本发明实施例提供了叠氮三氟甲氧基化合物及其合成方法,其中叠氮三氟甲氧基化合物的结构如下式(1)或式(2)所示:本发明提供的叠氮三氟甲氧基化合物,同时具有叠氮基和三氟甲氧基,能够通过进一步转化得到各种含氮官能团的三氟甲氧基化合物,可以做为医药、农药、材料等中间体。另外,本发明提供了通过温和的光催化的反应条件提供了一种能简单方便的合成叠氮三氟甲氧基化合物的新方法,底物使用范围广,无论底物是含吸电子还是给电子的乙烯类底物,都能得到较好的产率,并且该方法能够很好的适用于复杂天然产物分子的最后一步叠氮三氟甲氧基官能团化,应用范围广泛。
权利要求
1.叠氮三氟甲氧基化合物,其结构如下式(1)或式(2)所示:
其中,环A1、A2分别表示芳基或杂芳基,所述芳基或杂芳基可任选地被一至多个独立的R1取代;
Z表示-O-、-N-或-(CH
R1表示羟基、氨基、羧基、氰基、硝基、卤素、芳基、杂芳基、C
所述芳基或杂芳基的成环C原子可任选地被氧化为C(O)。
2.如权利要求1所述的叠氮三氟甲氧基化合物,其中环A1、A2分别表示6-14元芳基或6-14元杂芳基,所述芳基或杂芳基可任选地被1-3个独立的R1取代;
Z表示-O-、-N-或-(CH
R1表示氰基、卤素、6-14元芳基、6-14元杂芳基、C
所述芳基或杂芳基的成环C原子可任选被氧化为C(O)。
3.如权利要求2所述的叠氮三氟甲氧基化合物,其中:
所述芳基为苯基、萘基或
所述杂芳基为:苯并噻吩、苯并吡咯或苯并吡啶;
Z表示-(CH
R1表示氰基、卤素、苯基、甲基、叔丁基、甲氧基、溴代乙基氧基、苯基氧基、乙酰氧基、溴代乙酰氧基、甲磺酰基、甲氧基羰基、
4.如权利要求1-3中任一项所述的叠氮三氟甲氧基化合物,其选自如下结构的化合物:
5.如权利要求1-4中任一项所述的叠氮三氟甲氧基化合物的合成方法,其中,所述方法包括:在溶剂中,在光敏剂、银盐的存在下,优选溶剂中存在银盐配体的情况下,式(3)所示的化合物与叠氮化试剂及三氟甲氧基化试剂在惰性气氛中光照反应,得到式(1)所示的叠氮三氟甲氧基化合物;
或
式(4)所示的化合物与叠氮化试剂及三氟甲氧基化试剂在惰性气氛中光照反应,得到式(2)所示的叠氮三氟甲氧基化合物;
6.如权利要求5所述的叠氮三氟甲氧基化合物的合成方法,其中三氟甲氧基化试剂的结构如下式(5)所示;
式中,环A3表示苯基或萘基,所述苯基或萘基可任选地被1-3个独立的R2取代;
R2表示C
优选地,三氟甲氧基化试剂选自如下结构的化合物:
7.如权利要求5所述的叠氮三氟甲氧基化合物的合成方法,其中所述溶剂选自:二氯甲烷、乙酸乙酯、碳酸二甲酯、1,4-二氧六环、四氢呋喃、甲苯、乙腈、N,N-二甲基乙酰胺中的一种;优选选自乙腈分别与二氯甲烷、乙酸乙酯、碳酸二甲酯、1,4-二氧六环、四氢呋喃、甲苯、N,N-二甲基乙酰胺的组合。
8.如权利要求5所述的叠氮三氟甲氧基化合物的合成方法,其中所述光敏剂选自:[Ru(bpy)
[Ru(phen)
9.如权利要求5所述的叠氮三氟甲氧基化合物的合成方法,其中所述银盐选自:AgF、AgF
10.如权利要求5所述的叠氮三氟甲氧基化合物的合成方法,其中所述银盐配体选自:4,4',4”-三-叔丁基-2,2':6',2”-三联吡啶、
11.如权利要求5所述的叠氮三氟甲氧基化合物的合成方法,其中光照为蓝色光照射;优选为400-500nm波长的光照射。
12.如权利要求5-11中任一项所述的叠氮三氟甲氧基化合物的合成方法,其中所述叠氮化试剂为1-叠氮基-1,3苯并[d][1,2]碘羟-3(1H)-酮。
说明书
技术领域
本发明涉及有机合成技术领域,特别是涉及叠氮三氟甲氧基化合物及其合成方法。
背景技术
有机叠氮化物是含N结构基元的潜在前体,是重要的组成部分和中间体化合物。其显着的生物活性在药物化学、超分子化学、生物技术和材料科学等领域得到了重要的关注。而氟化有机化合物在药物,农药和材料中变得越来越重要,由于三氟甲氧基的独特性质,它们的强吸电性和高亲脂性,三氟甲氧基在新型农药中的引入备受关注,带有三氟甲氧基的分子将在各个领域得到应用,特别是在生命科学领域。合成该类同时具有叠氮和三氟甲氧基的官能团化合物具有者重要的意义。
到目前为止,烯烃类双键的三组分叠氮还是一个挑战。Studer等人在2013年描述了在TEMPONa存在下也使用高碘试剂的烯烃叠氮化物,然而,它们的方法需要使用钠金属新鲜制备的反应试剂TEMPONa,使得该反应操作局限性大,并且还特别引入四甲基哌啶氧化物基团(TEMPO),合成的产物比较单一(Stereoselective Radical Azidooxygenation ofAlkenes,Bo Zhang and Armido Studer*,Org.Lett.2013,15,4548–4551.)。在2014年,Jiao等人报道了使用铜催化剂,用高碘试剂(1-叠氮基-1,3苯并[d][1,2]碘羟-3(1H)-酮)吲哚氧基叠氮化,该反应的底物限制较大,只限制于β位有位阻的吲哚类底物(Copper-Catalyzed Oxoazidation and Alkoxyazidation of Indoles,Hang Yin,Teng Wang,andNing Jiao*Org.Lett.2014,16,2302-2305)。2015年,Greaney开发了一种新型的三组分铜催化还原偶联苯乙烯与亲核试剂如醇,NaN3,但是在这种方法中,他们使用了溶剂量的亲核试剂,NaN3也大大过量,且NaN3是十分危险的,反应的收率也不是很好(Three-ComponentAzidationofS tyrene-Type Double Bonds:Light-Switchable Behavior of aCopperPhotoredox Catalyst,Gabriele Fumagalli,Pauline T.G.Rabet,Scott Boyd,andMichael F.Greaney*,Angew.C hem.Int.E d.2015,54,1 1481–1148 4)。
目前为止,还没有人实现一步反应构建同时具有叠氮和三氟甲氧基的官能团化合物。
发明内容
本发明的目的在于提供一种叠氮三氟甲氧基化合物,同时,本发明还提供了叠氮三氟甲氧基化合物的合成方法,以实现通过一步反应构建同时具有叠氮和三氟甲氧基的官能团化合物。具体技术方案如下:
本发明首先提供了一种叠氮三氟甲氧基化合物,其结构如下式(1)或式(2)所示:
其中,环A1、A2分别表示芳基或杂芳基,所述芳基或杂芳基可任选地被一至多个独立的R1取代;
Z表示-O-、-N-或-(CH2)n-,其中n=1或2;
R1表示羟基、氨基、羧基、氰基、硝基、卤素、芳基、杂芳基、C1-6烷基、C2-6烯基、C2-6炔基、C1-6烷氧基、卤代C1-6烷氧基、芳基氧基、C1-6烷酰氧基、卤代C1-6烷酰氧基、C1-6烷基磺酰基、C1-6烷氧基羰基、
所述芳基或杂芳基的成环C原子可任选地被氧化为C(O)。
在本发明一些实施方式中,环A1、A2分别表示6-14元芳基或6-14元杂芳基,所述芳基或杂芳基可任选地被1-3个独立的R1取代;
Z表示-O-、-N-或-(CH2)n-,其中n=1-2;
R1表示氰基、卤素、6-14元芳基、6-14元杂芳基、C1-4烷基、C1-4烷氧基、卤代C1-4烷氧基、6-14元芳基氧基、C1-4烷酰氧基、卤代C1-4烷酰氧基、C1-4烷基磺酰基、C1-4烷氧基羰基、
所述芳基或杂芳基的成环C原子可任选被氧化为C(O)。
在本发明一些实施方式中:
所述芳基为苯基、萘基或
所述杂芳基为:苯并噻吩、苯并吡咯或苯并吡啶;
Z表示-(CH2)-;
R1表示氰基、卤素、苯基、甲基、叔丁基、甲氧基、溴代乙基氧基、苯基氧基、乙酰氧基、溴代乙酰氧基、甲磺酰基、甲氧基羰基、
在本发明一些实施方式中,式(1)所示的化合物其选自如下结构的化合物:
式(2)所示的化合物选自:
本发明还提供了前述叠氮三氟甲氧基化合物的合成方法,其中,所述方法包括:在溶剂中,在光敏剂、银盐的存在下,优选溶剂中存在银盐配体的情况下,式(3)所示的化合物与叠氮化试剂及三氟甲氧基化试剂在惰性气氛中光照反应,得到式(1)所示的叠氮三氟甲氧基化合物;
或
式(4)所示的化合物与叠氮化试剂及三氟甲氧基化试剂在惰性气氛中光照反应,得到式(2)所示的叠氮三氟甲氧基化合物。其中,环A1、A2及Z的定义如前所述。
本发明通过使用光敏剂(光氧化还原催化剂)首次结合金属银共同对反应进行催化循环;首先光敏剂对叠氮化试剂(例如,高价碘试剂)进行单电子还原,从而产生了叠氮自由基,使其与式(3)或式(4)所示的底物中的双键反应,得到底物自由基中间体,后光敏剂和一价银单电子转移,得到二价银,该二价银去进一步氧化底物自由基中间体,得到碳正离子,之后用三氟甲氧基化试剂在原位产生三氟甲氧基阴离子去捕获得到的碳正离子中间体,最后得到式(1)或式(2)所示的叠氮三氟甲氧基化合物。
在本发明的一些具体实施方式中,本发明提供的方法可以包括:在惰性气体,例如N2的保护下,向密封管中加光敏剂、银盐、叠氮化试剂、优选加入银盐配体,然后加入溶剂,将式(3)或式(4)所示的底物,三氟甲氧基化试剂加入到密封管中,然后将密封管中的混合物在室温并在光照射下搅拌2-6小时。之后,将反应混合物真空浓缩,通过硅胶色谱纯化,用石油醚/乙酸乙酯洗脱,得到式(1)或式(2)所示的叠氮三氟甲氧基化合物。
在本发明的一些具体实施方式中,三氟甲氧基化试剂与底物(式(3)或式(4)所示的化合物)的摩尔比为(2-10):1;优选为5:1;叠氮化试剂与底物的摩尔比为(1-3):1;优选为2:1;光敏剂与底物的摩尔比为(0.005-0.025):1;优选为0.01:1;银盐与底物的摩尔比为(0.25-0.75):1;优选为0.5:1;银盐配体与底物的摩尔比为(0.1-0.5):1;优选为0.3:1。
在本发明的一些具体实施方式中,
三氟甲氧基化试剂的结构如下式(5)所示;
式中,环A3表示苯基或萘基,所述苯基或萘基可任选地被1-3个独立的R2取代;
R2表示C1-4烷氧基羰基、C1-4烷基、C1-4烷氧基、硝基、卤素或卤代C1-4烷基;
优选地,三氟甲氧基化试剂选自如下结构的化合物:
在本发明的一些具体实施方式中,所述溶剂选自:二氯甲烷、乙酸乙酯、碳酸二甲酯、1,4-二氧六环、四氢呋喃、甲苯、乙腈、N,N-二甲基乙酰胺中的一种;优选选自乙腈,或优选选自乙腈分别与二氯甲烷、乙酸乙酯、碳酸二甲酯、1,4-二氧六环、四氢呋喃、甲苯、N,N-二甲基乙酰胺的组合。
在本发明的一些具体实施方式中,所述光敏剂选自:[Ru(bpy)3](PF6)2、[Ir{dF(CF3)ppy}2(dtbbpy)]PF6、[Ru(phen)3](PF6)2、Cu(dap)2Cl、[Ir(ppy)2(dtbbpy)](PF6)2、fac-Ir(ppy)3中的一种或其组合;优选选自:[Ru(bpy)3](PF6)2、[Ru(bpy)3](PF6)2或[Ru(phen)3](PF6)2。本发明所采用的各光敏剂可通过商业途径获得,或根据现有技术来制备;各光敏剂的分子式如表1所示。
表1 各光敏剂的分子式
在本发明的一些具体实施方式中,所述银盐选自:AgF、AgF2、Ag2O、AgO、Ag2CO3、AgBF4、Ag2SO4、AgSbF6、AgOAc中的一种或其组合;优选选自:AgF、AgF2、Ag2O或AgO。银盐与光敏剂相互作用,进行了单电子转移,使一价银转化成二价银。各银盐均市售可得。
在本发明的一些具体实施方式中,所述银盐配体选自:4,4',4”-三-叔丁基-2,2':6',2”-三联吡啶 中的一种或其组合;优选选自:4,4',4”-三-叔丁基-2,2':6',2”-三联吡啶。在反应过程中,银盐与银盐配体进行配位,降低银的氧化还原电势。
在本发明的一些具体实施方式中,光照为蓝色光照射;优选为400-500nm波长的光照射。
在本发明的一些具体实施方式中,叠氮化试剂为1-叠氮基-1,3苯并[d][1,2]碘羟-3(1H)-酮
本文中,术语“卤素”是指氟、氯、溴和碘。
本文中,术语“C1-6烷基”是指含有1-6个碳原子的直链或支链的饱和烃基,包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基和正已烷基。
本文中,术语“C2-6烯基”是指含有2-6个碳原子,且具有一个或多个碳碳双键的直链或支链的烃基,包括但不限于乙烯基、2-丙烯基和3-已烯基。
本文中,术语“C2-6炔基”是指含有2-6个碳原子,且具有一个或多个碳碳三键的直链或支链的烃基,还可以任选地包括一个或多个碳碳双键,包括但不限于乙炔基、丙炔基和3-已炔基。
本文中,术语“C1-6烷氧基”是指前文所定义的“C1-6烷基”通过氧原子与母体连接的基团,即“C1-6烷基-O-”基团,如甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、叔丁氧基、正戊氧基、新戊氧基和正己氧基等。所述的“C1-4烷氧基”指含有1-4个碳原子的上述实例,即“C1-4烷基-O-”基团。
本文中,术语“C1-6烷酰氧基”、“C1-6烷基磺酰基”及“C1-6烷氧基羰基”分别指:C1-6烷基-C(O)-O-、C1-6烷基-S(O)2-、C1-6烷基-O-C(O)-;所述“C1-6烷基”如前文所定义,优选为“C1-4烷基”;术语“芳基氧基”指:芳基-O-。
本文中,术语“卤代C1-6烷氧基”及“卤代C1-6烷酰氧基”是指该基团的C1-6烷基中的氢原子被一个或多个卤素取代;所述“卤素”如前文所定义。
本文中,术语“芳基”及“杂芳基”,包括单环系统和稠环系统(双环系统或者多环系统),单环是指仅以一个环的形式存在,稠环是指两个及两个以上环以并、螺、桥的连接方式所形成的多环系结构。所述的并环是指由两个或两个以上环状结构彼此共用两个相邻的环原子(即共用一个键)所形成的稠环结构。所述的桥环是指由两个或两个以上环状结构彼此共用两个非相邻的环原子所形成的稠环结构。所述的螺环是指由两个或两个以上环状结构彼此共用一个环原子所形成的稠环结构。本发明以原子个数所限定的芳基、杂芳基,在不特别指明的情况下,包括所能够形成的单环和稠环结构。
本文中,术语“芳基”,是指芳香性的环状基团,包括指单环系统、双环系统或多环系统,可以为6-14元芳基,包括苯基、萘基、菲基等。
本文所用的术语“杂芳基”是指至少一个环碳原子被选自O、S、N的杂原子替代的芳香性的环状基团,优选1-3个杂原子,同时包括碳原子、硫原子被氧代的情况,例如碳原子被C(O)、S(O)、S(O)2替代。杂芳基包括单杂芳基和稠杂芳基,单杂芳基的代表性实例包括但不仅限于:呋喃基、咪唑基、异恶唑基、噻唑基、苯并噻吩基、苯并吡咯基、苯并吡啶基等。
本文中,术语“被……取代”意指给定原子或基团上的一个或多个氢原子被一个或多个选自给定的取代基替换,条件是不超过该给定原子的正常化合价。
本文中,术语“被一个或多个取代基取代”意指给定的原子或基团上的一个或多个氢原子独立地被一个或多个选自给定基团的取代基替换。
缩写
本文中所涉及的缩写如下所示,对于文中涉及但未列出的缩写,其具有本领域的通常含义。
Ac乙酰基
Ac2O乙酸酐
BF3·Et2O 三氟化硼-二乙醚
Boc 叔丁氧基羰基
DMA N,N-二甲基乙酰胺
DMAP4-二甲氨基吡啶
DCM 二氯甲烷
DMC 碳酸二甲酯
DMF N,N-二甲基甲酰胺
DMSO二甲基亚砜
EA乙酸乙酯
Et2O二乙醚
EDCI1-(3-二甲基氨基丙基)-3-乙基碳二亚胺
EtOAc 乙酸乙酯
Et3N三乙胺
Me甲基
MeCN乙腈
Ms甲烷磺酰基(甲磺酰基)
n-BuLi正丁基锂
PE石油醚
Ph苯基
Py吡啶
t-Bu叔丁基
THF 四氢呋喃
Ts对甲苯磺酰基
本发明提供的叠氮三氟甲氧基化合物,同时具有叠氮基和三氟甲氧基,能够通过进一步转化得到各种含氮官能团的三氟甲氧基化合物,可以做为医药、农药、材料等中间体。另外,本发明提供了一种在温和的光催化的反应条件下合成叠氮三氟甲氧基化合物的新方法,该方法适于的底物范围广,无论底物是含吸电子还是给电子的含碳碳双键的底物,都能得到较好的产率,并且该方法能够很好的适用于复杂天然产物分子的最后一步叠氮三氟甲氧基官能团化,应用范围广泛。
具体实施方式
本发明以式(3)或式(4)所示的含碳碳双键的化合物作为底物进行反应,下述各具体实施例中,式(3)或式(4)所示的化合物可以通过商业途径购得,或可以根据现有技术的记载而合成;下面以示例的方式对部分底物的合成进行较为详细的说明。
底物的合成实施例
实施例1
在氮气下,在氮气氛下,将甲基三苯基溴化鏻(0.940g,2.70mmol,1.20当量)悬浮于THF(6.00mL)中并且逐滴添加正丁基锂(1.20mL,2.40M的THF溶液,2.70mmol,1.20当量)在-78℃搅拌反应1h后,在-78℃滴加4-(2-溴乙氧基)苯甲醛(0.500g,2.20mmol,1.00当量)在THF(2.00mL)中的溶液。然后,反应在30℃下搅拌12小时。用H2O(5.00mL)淬灭反应并用乙醚(5.00mL)萃取3次。合并的有机层用MgSO4干燥。将滤液真空浓缩,残余物经硅胶层析纯化,用己烷/二氯甲烷(DCM)30:1(v/v)洗脱,得到0.950g1-(2-溴乙氧基)-4-乙烯基苯(1l),为白色固体(产率93%)。Rf=0.4(hexanes/DCM 30:1(v/v)).NMR Spectroscopy:
实施例2
向搅拌的4-羟基苯乙烯(0.60g,5.00mmol,1.00当量)的DCM(20.0mL)溶液中缓慢加入吡啶(0.60mL,7.50mmol,1.50当量)和2-溴乙酰溴(0.48mL,5.50mmol),并将该混合物在0℃下搅拌15分钟,再在室温下再搅拌20分钟。将水加入到溶液中,并将混合物用DCM萃取。将合并的有机层用盐水洗涤,用Na2SO4干燥,真空浓缩。通过使用PE/EtOAc(v/v=20/1)作为洗脱剂的快速硅胶柱色谱进行纯化,得到525mg 4-乙烯基苯基2-溴乙酸酯(1k)(44%产率),为乳白色固体。
实施例3
在50mL干燥的Schelenk烧瓶中,向Gibberellic acid(反应物)(880mg,2.54mmol,1.00当量)的30ml DCM溶液中依次加入Et3N(2.10ml,15.24mmol,6.0当量),EDCI(974mg,5.08mmol,2.00在冰水浴中加入DMAP(62mg,0.508mmol,0.20当量)和4-羟基乙烯(458mg,3.81mmol,1.5当量)。将反应混合物缓慢温热至室温并搅拌24小时。然后向混合物中加入Et3N(10.5ml,76.2mmol,30.0当量)和Ac2O(4.00ml,4.26mmol,16.70当量)。4小时后,用冰水浴中的20ml水淬灭反应,用DCM(15mL×3)萃取,用MgSO4干燥并过滤。将滤液真空浓缩。通过SiO2快速柱色谱纯化残余物,用己烷/EtOAc 4:1至3:1(v/v)洗脱,得到500mg产物(37%,白色固体)
实施例4
将三氟化硼-二乙醚(3滴)加入到二乙醚(30mL)中的DHA(426mg,1.50mmol,1.00当量)和(4-乙烯基苯基)甲醇(370mg,2.76mmol,1.84当量)毫升)。6小时后,反应混合物用饱和NaHCO 3水溶液淬灭并干燥(MgSO4)。过滤并浓缩滤液,得到残余物,用乙酸乙酯/己烷(1:10)层析,得到产物,为无色油状物(198mg,33%)。
实施例5
Ac-齐墩果酸(Ac-oleanic acid)的合成:在25mL干燥的Schelenk烧瓶中,在冰水浴中,在10mL无水吡啶中的齐墩果酸(oleanic acid,2.00mmol,1.00当量)中加入Ac2O(1.50当量)。将反应混合物缓慢温热至室温(rt)过夜。反应混合物用水洗涤数次,用乙酸乙酯萃取,然后用MgSO4干燥。滤液真空浓缩,得到纯的Ac-齐墩果酸,用于下一步(>99%)。
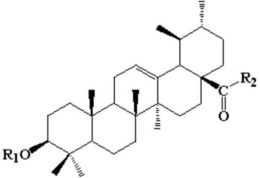
Ac-齐墩果酸酯(Ac-oleanic acid ester)的合成:在25mL干燥的Schelenk烧瓶中,向在5.00mL无水DCM中的Ac-齐墩果酸(698mg,1.40mmol,1.00当量)缓慢地加入草酰氯(Oxalyl chloride,2.00当量)(一滴DMF加入)。将反应混合物在室温下搅拌3小时。将溶剂真空除去,得到Ac-齐墩果酰氯。然后,在室温下,向10.0mL无水DCM中的4-乙烯基苯酚(2.80mmol,2.00当量),Et3N(2.80mmol,2.00当量)和DMAP(5mol%)的溶液中缓慢加入Ac-齐墩果酰氯。得到的混合物在N 2下回流12小时。用水洗涤并用DCM(5.00mL×3)萃取。合并的DCM相经MgSO4干燥,真空除去溶剂。通过SiO2快速柱色谱法用洗脱剂己烷/EtOAc 3:1(v/v)纯化残余物,得到644mg Ac-齐墩果酸酯(1dd)(75%),白色固体。
NMR Spectroscopy:
实施例1-5中的产率均是指分离产率;分离产率的计算方法为本领域技术人员所公知,本发明在此不再进行限定。
叠氮三氟甲氧基化合物的合成实施例
实施例6
1-(2-叠氮基-1-(三氟甲氧基)乙基)-2,4,6-三甲基苯(4a)
在N2保护下,向密封管中加Ru(bpy)3(PF6)2(2.4mg,0.00300mmol,1.0mol%),AgF(19.2mg,0.150mmol,50.0mol%),4,4',4”-三-叔丁基-2,2':6',2”-三联吡啶(tbtpy,36.0mg,0.0900mmol,30.0mol%),1-叠氮基-1,3苯并[d][1,2]碘羟-3(1H)-酮(3)(173.4mg,0.600mmol,2.00当量),然后加入乙腈溶剂(2.40mL),4-苯基苯苯乙烯(1a)(43.9mg,0.300mmol,1.00当量),三氟甲基4-甲基苯磺酸酯(2)(169μL,0.9mmol,3.00equiv)加入到反应中,然后将所得混合物至于室温并在14W蓝色LED的照射下搅拌4小时。之后,将反应混合物真空浓缩,通过硅胶色谱纯化,用石油醚/乙酸乙酯100:1(v/v)洗脱,得到相应产物(4a),65.5mg。
实施例7-21
将实施例6的溶剂替换成其它不同的溶剂后,完成实施例7-21,实施例7-21所采用的溶剂见表2,同时实施例6-21产物的产率记录于表2中。
表2
实施例22-27
将实施例6的光敏剂用量改变后完成实施例22,将实施例6的光敏剂替换成其它不同的光敏剂后,完成实施例23-27,实施例22-27所采用的光敏剂见表3,同时实施例22-27产物的产率记录于表3中。
表3
实施例28-35
将实施例6的银盐替换成其它不同的银盐后,完成实施例28-35,实施例28-35所采用的银盐见表4,同时实施例28-35产物的产率记录于表4中。
表4
实施例36-43
将实施例6的三氟甲基4-甲基苯磺酸酯的用量改为1.5mmol后,完成实施例36,将实施例6的三氟甲基4-甲基苯磺酸酯替换成其它不同的三氟甲氧基化试剂(用量1.5mmol)后,完成实施例37-43,实施例37-43所采用的三氟甲氧基化试剂见表5,同时实施例36-43产物的产率记录于表5中。
表5
实施例44-50
将实施例36的银盐配体去掉或替换成其它不同的银盐配体(用量不变)后,完成实施例44-50,实施例44-50所采用的银盐配体见表6,同时实施例44-50产物的产率记录于表6中。
表6
实施例51-81
将实施例36的底物替换为其它含碳碳双键的化合物后,按照实施例36的合成过程完成实施例51-81;所得的产物及产率(分离产率)见表7。
表7
其中:
实施例52的底物可以根据文献Appukkuttan.P.,Dehaen.W.,Eycken.E.V.Chem.Eur.J.2007,13,6452–6460.来合成。
实施例53的底物可以根据文献Gülak.S.,Gieshoff.T.,Wangelin.A.J.Adv.Synth.Catal.2013,355,2197–2202.来合成。
实施例54的底物可以根据文献Greenhalgh.M.D.,Thomas,S.P.J.Am.Chem.Soc.2012,134,11900-11903.来合成。
实施例56的底物可以根据文献Hu.W.X.,Li.P.R.,Jiang.J.X.,Che.C.M..,Chen.X.Adv.Synth.Catal.2010,11,3190–3194.来合成。
实施例58的底物可以根据文献Liu.G.,D.Org.Lett.2009,11,3670-3673.来合成。
实施例74的底物可以根据文献Falk.A.,Cavalieri.,Nichol.G.S.,Vogt.D.,Schmalzb.H.G.Adv.Synth.Catal.2015,357,3317–3320.来合成。
实施例75的底物可以根据文献Falk.A.,Cavalieri.,Nichol.G.S.,Vogt.D.,Schmalzb.H.G.Adv.Synth.Catal.2015,357,3317–3320.来合成。
实施例77的底物可以根据文献Guo.S.,Cong.F.,Guo.R.,Wang L.,Tang.P.P.Nat.Chem.,2017,9,546-551.来合成。
实施例78的底物可以根据文献Guo.S.,Cong.F.,Guo.R.,Wang L.,Tang.P.P.Nat.Chem.,2017,9,546-551.来合成。
表7中其它实施例的底物均可以从商业途径获得,本发明在此不再赘述。
应用实施例
通常来说,含叠氮的化合物能够很容易的实现多种官能团的转化反应。本发明提供的叠氮三氟甲氧基化合物可以实现,例如但不限于:
a)通过叠氮基团进行点击化学反应,得到含三氟甲氧基的化合物,这在生物化学蛋白标记,以及活性药物分子衍生化等方面有很重要的意义。下面以叠氮三氟甲氧基化合物(4a)为例进行说明。
实施例82
在10ml圆底烧瓶中,将1-(2-叠氮基-1-(三氟甲氧基)乙基)-4-苯基苯(4a)(230mg,0.75mmol)和苯乙炔(81mg,1.05当量)在25℃下溶解于H2O(1mL)中的硫酸铜(II)五水合物(9.4mg,0.05当量),抗坏血酸钠(sodium ascorbate,23mg,0.15当量)和β-环糊精(β-CD,22mg,0.025当量)的混合物。将反应混合物在室温下搅拌50分钟。将所得混合物倒入DCM(3mL)和H2O(3mL)中,分离有机层。将水层用DCM(3×3mL)萃取。将合并的有机层真空浓缩。通过硅胶短柱色谱纯化残余物,用己烷/EtOAc 2:1(v/v)洗脱,得到292mg的5(95%收率)。Rf=0.1hexanes/EtOAc 8:1(v/v)).NMRSpectroscopy:
b)本发明提供的叠氮三氟甲氧基化合物还可以通过连续三步的反应(中间产物不用分离),就能够很好地得到药物Midodrine的含氧三氟甲基的生物电子等排体。下面以实施例54的产物2-(2-叠氮基-1-(三氟甲氧基)乙基)-1,4-二甲氧基苯为例进行说明。
实施例83
将2-(2-叠氮基-1-(三氟甲氧基)乙基)-1,4-二甲氧基苯(4e)(40.0mg,0.136mmol,1.00当量),10%Pt/C(4.0mg,0.0136mmol)在EtOH(2.00ml)中的混合物在氢气氛下于25℃剧烈搅拌8小时。用硅藻土过滤后,真空蒸发溶剂。将粗产物(主要产物的Rf=0.1DCM/MeOH 10:1(v/v))不需要进一步纯化,溶于乙酸乙酯(5ml)中。在另一个烧瓶中,将1,1'-羰基二咪唑(CDI,97.0mg,0.600mmol,4.40当量)悬浮于乙酸乙酯(2.00ml)中。向米色悬浮液中加入N-叔丁氧基羰基甘氨酸(Boc-glycin,105mg,0.600mmol,4.40当量)。搅拌1小时后,将该溶液加入到上述溶液中。将反应混合物在室温下搅拌1小时。将1M HCl(3.00ml)的溶液加入到反应混合物中,并将混合物在室温下搅拌15分钟。搅拌停止,各相分离。依次用水,2.5%氢氧化钠和水洗涤有机层,然后用硫酸钠干燥。向澄清的乙酸乙酯溶液中通入HCl(气体)30分钟,并将白色悬浮液在室温下搅拌1小时。将混合物真空浓缩。然后将残余物溶于乙酸乙酯中。乙酸乙酯溶液依次用饱和碳酸钠和盐水洗涤,然后用硫酸钠干燥并真空浓缩。通过硅胶快速色谱法(DCM/MeOH 10:1(v/v))纯化残余物,得到15.0mg2-氨基-N-(2-(2,5-二甲氧基苯基)-2-(三氟甲氧基)乙基)乙酰胺(8)。Rf=0.1DCM/MeOH 10:2(v/v).
以上对本发明所提供的叠氮三氟甲氧基化合物及其合成方法进行了详细介绍。本文中应用了具体实施例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其中心思想。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围。
叠氮三氟甲氧基化合物及其合成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0