

IPC分类号 : C07J43/00,A61K31/58,A61P3/10,A61P3/06,A61P9/12,A61P9/10,A61P3/04,A61P3/00








专利摘要
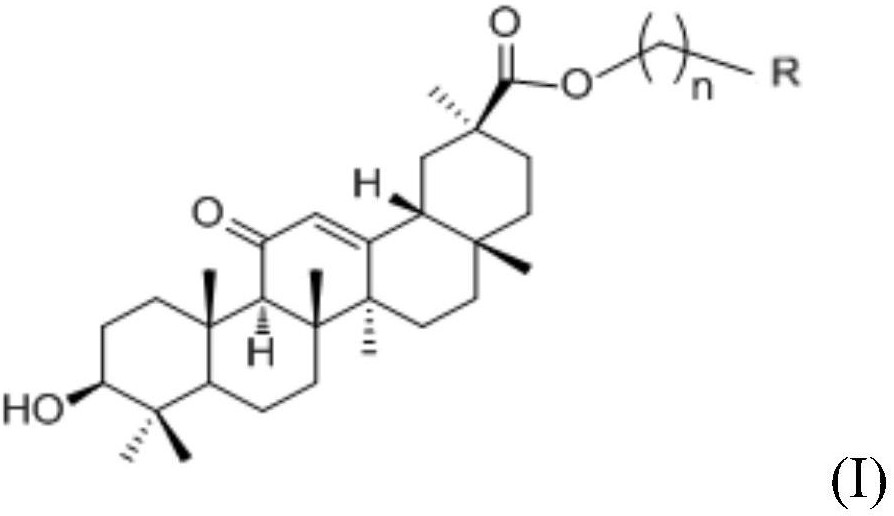
本发明属于药物合成技术领域,公开了一种糖原磷酸化酶抑制剂及其制备方法和应用。所述糖原磷酸化酶抑制剂具有式(I)所示的结构通式或其药学上可接受的盐或酯,其中X1、X2、X3和X4均为碳原子连接基团或者X1、X2、X3和X4其中之一为氮原子连接基团而其他均为碳原子连接基团;R1为H、卤素、羟基、氰基、C1‑6的烷基、C1‑6的烷氧基、三氟甲基、乙烯基或乙炔基。本发明具有新型结构的糖原磷酸化酶抑制剂对糖原磷酸化酶具有抑制作用,且其在细胞水平的抑制糖原分解作用与PSN‑357相当。可用于制备预防和治疗糖尿病及其并发症、高血脂症、高血压及其并发症、动脉粥样硬化症、肥胖、代谢综合症药物。
权利要求
1.一种糖原磷酸化酶抑制剂,其特征在于:所述糖原磷酸化酶抑制剂具有式(I)所示的结构通式或其药学上可接受的盐或酯:
其中:X1、X2、X3和X4均为碳原子连接基团或者X1、X2、X3和X4其中之一为氮原子连接基团而其他均为碳原子连接基团;R1为H、卤素、羟基、氰基、C1-6的烷基、C1-6的烷氧基、三氟甲基、乙烯基或乙炔基。
2.根据权利要求1所述的一种糖原磷酸化酶抑制剂,其特征在于:所述R1为H、卤素或氰基。
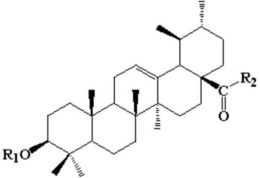
3.根据权利要求1所述的一种糖原磷酸化酶抑制剂,其特征在于:所述糖原磷酸化酶抑制剂具有如下式(I1)所示的结构式或其药学上可接受的盐或酯:
4.权利要求1或2所述的一种糖原磷酸化酶抑制剂的制备方法,其特征在于包括如下制备步骤:
(1)以式1结构的化合物为起始原料,经氢化还原、氧化、叠氮化获得式2结构的化合物;
(2)式2结构的化合物在DCC和DMAP催化下,与具有式3结构的化合物进行酯化反应,得到具有式4结构的酯化产物,然后经三氟乙酸脱保护剂得到式5结构的含游离羧基化合物;
(3)式5结构的化合物在HATU和DIPEA催化下,与式6结构的胆酸衍生物经酰化反应得到具有式(I)结构的目标化合物糖原磷酸化酶抑制剂;
5.根据权利要求4所述的一种糖原磷酸化酶抑制剂的制备方法,其特征在于:步骤(1)中所述的氢化还原是指在碱性条件下采用Pd/C催化剂进行氢化还原;所述氧化是指采用过硫酸氢钾复合盐在水和二氯甲烷的混合溶剂中的氧化;所述的叠氮化是指在AcOH溶剂下用4-氨基苯甲酸叔丁酯进行叠氮化。
6.根据权利要求4所述的一种糖原磷酸化酶抑制剂的制备方法,其特征在于:所述具有式6结构的胆酸衍生物通过如下方法制备:
将胆酸溶解于无水DMF中,然后加入N-Boc-乙二胺、焦碳酸二乙酯和三乙胺,室温搅拌反应,反应产物经分离纯化,得白色固体产物N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-N’-叔丁氧羰基乙二胺;然后将其溶于甲醇中,冰浴下滴加新鲜制备的氯化氢的甲醇溶液脱保护,得到具有式6结构的胆酸衍生物N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-乙二胺。
7.权利要求1~3任一项所述的糖原磷酸化酶抑制剂在制备预防和治疗糖尿病及其并发症、高血脂症、高血压及其并发症、动脉粥样硬化症、肥胖、代谢综合症药物中的应用。
8.根据权利要求7所述的应用,其特征在于:所述药物包含作为活性剂的式(I)结构的化合物或其药学上可接受的盐或酯和药学上可接受的载体。
9.根据权利要求8所述的应用,其特征在于:所述药学上可接受的载体是指一种或几种惰性的、不逆向与活性剂或病人发生作用的、非毒性的固体或液体填充物、稀释剂或助剂。
10.根据权利要求7~9任一项所述的应用,其特征在于:所述药物的剂型为片剂、胶囊、丸剂、栓剂、口服液、混悬液或注射液。
说明书
技术领域
本发明属于药物合成技术领域,具体涉及一种糖原磷酸化酶抑制剂及其制备方法和应用。
背景技术
糖尿病是继肿瘤、心血管疾病之后第三大严重威胁人类健康的慢性非传染性疾病。目前我国有糖尿病患者9200多万,成为全球第一糖尿病大国,其中约97%患者属于2型糖尿病。对于2型糖尿病的治疗,根据药物药理作用机制的不同,分为胰岛素及其同类药物、胰岛素增敏剂、胰岛素促泌剂、减少碳水化合物调节剂等。但这些药物的降糖效果有限。随着人们对糖尿病发病机制认识的不断深入,许多治疗糖尿病的新型作用靶点被发现,新型药物不断问世。
糖原磷酸化酶(GP)为糖代谢途径中的重要酶,催化糖原降解的关键酶。抑制该酶的活性,可以减少糖原的降解,达到减少肝脏葡萄糖的生成,从而降低血糖的目的。近年来,GP抑制剂作为潜在的降血糖药物已受到广泛关注。其特点在于在降低血糖的同时,对缺血性心肌损伤有显著的保护作用,这一点对患有缺血性心血管并发症的糖尿病患者是尤其重要的。
US6297269和EP0832066报道了作为GP抑制剂的取代的N-(吲哚-2-羰基)酰胺和衍生物。US6107329和US6277877报道了作为GP抑制剂的取代的N-(吲哚-2-羰基)甘氨酰胺和衍生物。US6399601报道了双环吡咯酰胺类GP抑制剂。US5952322公开了GP抑制剂用于降低非心脏缺血性组织损伤的方法。
目前Pfizer的CP-368296和OSI公司的氮杂吲哚酰胺衍生物PSN-357已进入临床研究阶段。另外,也有一些天然的五环三萜类化合物具有通过抑制肝脏葡萄糖的生成达到降血糖的目的,分子模拟手段证实了这些五环三萜类化合物具有与GP可能的相互作用位点。
总之,GP与过量肝脏葡糖糖生成的调节相关,其抑制剂的降血糖作用将对联合治疗及其相关的心血管疾病的治疗是非常有利的。
发明内容
为了进一步开发安全有效的治疗糖尿病的新药物,本发明的首要目的在于提供一种糖原磷酸化酶抑制剂。
本发明的另一目的在于提供上述糖原磷酸化酶抑制剂的制备方法。
本发明的再一目的在于提供上述糖原磷酸化酶抑制剂在制备预防和治疗糖尿病及其并发症、高血脂症、高血压及其并发症、动脉粥样硬化症、肥胖、代谢综合症药物中的应用。
本发明目的通过以下技术方案实现:
一种糖原磷酸化酶抑制剂,所述糖原磷酸化酶抑制剂具有式(I)所示的结构通式或其药学上可接受的盐或酯:
其中:X1、X2、X3和X4均为碳原子连接基团或者X1、X2、X3和X4其中之一为氮原子连接基团而其他均为碳原子连接基团;R1为H、卤素、羟基、氰基、C1-6的烷基、C1-6的烷氧基、三氟甲基、乙烯基或乙炔基。
优选地,所述R1为H、卤素或氰基。
更优选地,所述糖原磷酸化酶抑制剂具有如下式(I1)所示的结构式或其药学上可接受的盐或酯:
上述糖原磷酸化酶抑制剂的制备方法,包括如下制备步骤:
(1)以式1结构的化合物(邻硝基苯基丙烯酸)为起始原料,经氢化还原、氧化、叠氮化获得式2结构的化合物((E)-3-(2-((4-(叔丁氧羰基)苯基)叠氮基)苯基)丙酸);
(2)式2结构的化合物在DCC(二环己基碳二亚胺)和DMAP(4-二甲氨基吡啶)催化下,与具有式3结构的化合物进行酯化反应,得到具有式4结构的酯化产物,然后经三氟乙酸脱保护剂得到式5结构的含游离羧基化合物;
(3)式5结构的化合物在HATU(2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯)和DIPEA(N,N-二异丙基乙胺)催化下,与式6结构的胆酸衍生物经酰化反应得到具有式(I)结构的目标化合物糖原磷酸化酶抑制剂;
上述制备方法的合成路线图如图1所示。
优选地,步骤(1)中所述的氢化还原是指在碱性条件下采用Pd/C催化剂进行氢化还原;所述氧化是指采用过硫酸氢钾复合盐(Oxone)在水和二氯甲烷的混合溶剂中的氧化;所述的叠氮化是指在AcOH溶剂下用4-氨基苯甲酸叔丁酯进行叠氮化。
上述具有式3结构的化合物可参考CN200480021117.2进行制备。
优选地,所述具有式6结构的胆酸衍生物通过如下方法制备:
将胆酸溶解于无水DMF中,然后加入N-Boc-乙二胺、焦碳酸二乙酯(DEPC)和三乙胺(Et3N),室温搅拌反应,反应产物经分离纯化,得白色固体产物N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-N’-叔丁氧羰基乙二胺;然后将其溶于甲醇中,冰浴下滴加新鲜制备的氯化氢的甲醇溶液脱保护,得到具有式6结构的胆酸衍生物N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-乙二胺。
上述糖原磷酸化酶抑制剂在制备预防和治疗糖尿病及其并发症、高血脂症、高血压及其并发症、动脉粥样硬化症、肥胖、代谢综合症药物中的应用。
上述应用中,所述药物包含作为活性剂的式(I)结构的化合物或其药学上可接受的盐或酯和药学上可接受的载体。
上述药学上可接受的载体是指药学领域常规的药物载体,是指一种或几种惰性的、非毒性的固体或液体填充物、稀释剂、助剂等,他们不逆向与活性化合物或病人发生作用。
上述药物的剂型可以是片剂、胶囊、丸剂、栓剂、口服液、混悬液、注射液等药剂学上常用的剂型。
上述片剂、丸剂和胶囊含有传统的赋形剂如填充物、稀释剂、润滑剂、分散剂以及粘合剂。上述各种剂型可以按照药学领域中熟知的方法制备。以上活性剂的剂量因配方而异。
本发明具有如下优点及有益效果:
本发明合成了一种新型结构的糖原磷酸化酶抑制剂。该抑制剂对糖原磷酸化酶具有抑制作用,且其在细胞水平的抑制糖原分解作用与PSN-357相当。因此可用于制备预防和治疗糖尿病及其并发症、高血脂症、高血压及其并发症、动脉粥样硬化症、肥胖、代谢综合症药物。
附图说明
图1为本发明所述糖原磷酸化酶抑制剂的合成路线图。
具体实施方式
下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
本实施例一种糖原磷酸化酶抑制剂的制备,具体制备步骤如下:
(1)(E)-3-{2-[(4-叔丁氧羰基苯基)叠氮基]苯基}丙酸的合成:
(E)-3-(硝基苯基)丙烯酸(10.0g,52.0mmol)溶于水(500mL),加入NaOH(4.1g,39mmol)和10%Pd/C(0.5g),室温常压氢化过夜。反应物过滤除去Pd/C,减压蒸去溶剂后得白色粉未状固体粗品(7.5g,87%)。此粗品溶于水(200mL)和二氯甲烷(300mL)的混合溶剂中,加入过硫酸氢钾复合盐(Oxone,55.3g,90.0mmol),室温搅拌2h。萃取收集二氯甲烷有机相,水相以二氯甲烷萃取,合并有机相,无水硫酸钠干燥后蒸去溶剂,得桔色固体粗品(7.5g,92.5%)。此粗品溶于AcOH(75.0mL),加入4-氨基苯甲酸叔丁酯(8.1g,42.0mmol),80℃搅拌16h。减压蒸除溶剂,残渣经快速柱层析(石油醚/乙酸乙酯:10/1,V/V)得桔色固体产物(3.5g,25%)。产物鉴定数据如下:
ESI-MS m/z:355.2[M+H]+;
1H NMR:(CDCl3,400MHz)8.15(d,J=8.4Hz,2H),7.93(d,J=8.4Hz,2H),7.72(d,J=7.6Hz,1H),7.40.7.47(m,2H),7.32-7.37(m,1H),3.49(t,J=7.6Hz,2H),2.79(t,J=8.0Hz,2H)。
(2)(E)-3-{2-[(4-叔丁氧羰基苯基)叠氮基]苯基}丙酸-{1-[2-(5-氯-1H-吡咯并[2,3-c]吡啶-2-甲酰胺)-3-(4-氟苯基)丙酰基]-哌啶-4-基}酯的合成:
(E)-3-{2-[(4-叔丁氧羰基苯基)叠氮基]苯基}丙酸(240.0mg,0.68mmol),催化量的DMAP(8.3mg,0.068mmol)溶于二氯甲烷(8.0mL)中,冰浴下加入DCC(153.4mg,0.75mmol),冰浴下搅30min,再加入具有式7结构的化合物(300mg,0.68mmol,参考CN200480021117.2进行制备),室温搅拌过夜。反应混合物过滤,滤液蒸去溶剂,残余物用乙酸乙酯溶解,置冰箱中过夜,过滤,滤液蒸去溶剂,残渣快速柱层析(石油醚/乙酸乙酯:10/1,V/V)得桔色固体(440.0mg,82%)。产物鉴定数据如下:
ESI-MS m/z:781.2[M+H]+;
1H NMR:(MeOD,400MHz)8.49(d,J=9.6Hz,1H),8.07(dd,J=8.8,20.4Hz,2H),7.88(dd,J=8.8,20.8Hz,2H),7.63(dd,J=7.6,28.0Hz,1H),7.58(d,J=8.0Hz,1H),7.22-7.46(m,5H),7.07(d,J=5.6Hz,1H),6.92-7.00(m,2H),5.23(dd,J=7.2,11.6Hz,1H),4.76-4.85(m,1H),3.37-3.50(m,5H),2.98-3.22(m,3H),2.64-2.71(m,2H),1.82-1.86(m,2H),1.63-1.72(m,2H),1.58(d,J=9.6Hz,9H)。
(3)(E)-3-{2-[(4-苯甲酸)叠氮基]苯基}丙酸-{1-[2-(5-氯-1H-吡咯并[2,3-c]吡啶-2-甲酰胺)-3-(4-氟苯基)丙酰基]-哌啶-4-基}酯的合成:
(E)-3-{2-[(4-叔丁氧羰基苯基)叠氮基]苯基}丙酸-{1-[2-(5-氯-1H-吡咯并[2,3-c]吡啶-2-甲酰胺)-3-(4-氟苯基)丙酰基]-哌啶-4-基}酯(650.0mg,0.83mmol)溶于二氯甲烷(10mL),冰浴下滴加三氟醋酸(10mL,140mmol),滴毕维持此温度搅拌2h。减压蒸除反应液,残渣溶于乙酸乙酯,依次以饱和碳酸氢钠溶液和饱和食盐水洗涤,无水Na2SO4干燥。过滤除去干燥剂,浓缩,得到桔色固体粗品,此粗品无需纯化可直接用于下一步反应。产物鉴定数据如下:
ESI-MS m/z:725.1[M+H]+;
1H NMR:(CDCl3,400MHz)8.62(s,1H),8.22(d,J=13.6Hz,1H);8.20(d,J=14.0Hz,1H),7.99(d,J=20.0Hz,1H),7.96(d,J=20.0Hz,1H),7.75(d,J=2.4Hz,1H),7.70(dd,J=8.0,30.0Hz,1H),7.27-7.53(m,5H),7.10(s,1H),6.97-7.04(m,2H),5.27(t,J=7.2Hz,1H),4.79-4.85(m,1H),.41-3.70(m,5H),3.04-3.27(m,3H),2.69-2.79(m,2H),1.85-1.89(m,2H),1.69-1.79(m,2H)。
(4)N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-N’-叔丁氧羰基乙二胺的合成:
胆酸(2.0g,4.9mmol)溶解于无水DMF(20.0mL)中,缓慢加入N-Boc-乙二胺(784.3mg,4.9mmol),焦碳酸二乙酯(DEPC,879.0mg,5.4mmol)和三乙胺(Et3N,2.5g,5.0mmol),加毕,室温搅拌24h。反应混合物过滤,滤液蒸去溶剂,残渣快速柱层析(乙酸乙酯/四氢呋喃:10/1,V/V)得白色固体(2.0g,73%)。产物鉴定数据如下:
ESI-MS m/z:451.2[M+H]+;
1H NMR:(DMSO-d6,400MHz)7.73(t,J=5.2Hz,1H),6.73(t,J=5.6Hz,1H),4.28(br s,1H),4.06(s,1H),3.96-3.99(m,1H),3.74(s,1H),3.56(s,1H),3.11-3.17(m,1H),2.97-3.00(m,2H),2.89-2.92(m,2H),1.33(s,9H),0.87(d,J=6.4Hz,3H),0.76(s,3H),0.54(s,3H)。
(5)N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-乙二胺的合成:
N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-N’-叔丁氧羰基乙二胺(200.0mg,0.36mmol)溶于甲醇(20mL)中,冰浴下,缓慢滴加新鲜制备的氯化氢的甲醇溶液(2N,20mL),室温搅拌2h。过滤,滤饼以二氯甲烷洗涤,真空干燥得淡黄色固体(170mg,97%)。产物鉴定数据如下:
ESI-MS m/z:473.3[M+Na]+;
1H NMR:(DMSO-d6,400MHz)8.01-8.14(m,4H),3.74(br s,4H),3.56(br s,1H),3.24(dd,J=6.0,11.6Hz,2H),3.14-3.18(m,1H),2.78(dd,J=5.6,11.2Hz,2H),0.88(d,J=6.0Hz,2H),0.76(s,3H),0.54(s,3H)。
(6)(E)-3-[2-({4-[N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-乙二胺基]苯甲酰基}叠氮基)苯基]丙酸-{1-[2-(5-氯-1H-吡咯并[2,3-c]吡啶-2-甲酰胺)-3-(4-氟苯基)丙酰基]-哌啶-4-基}酯的合成:
(E)-3-{2-[(4-苯甲酰基)叠氮基]苯基}丙酸-{1-[2-(5-氯-1H-吡咯并[2,3-c]吡啶-2-甲酰胺)-3-(4-氟苯基)丙酰基]-哌啶-4-基}酯(150mg,0.207mmol)溶于DMF(3mL)中,冰浴下加入HATU(94mg,0.248mmol)和DIPEA(0.1mL,0.621mmol),室温搅拌10min,再加入N-(3α,7α,12α-三羟基-5β-胆烷酰胺)-乙二胺(120mg,0.266mmol),室温搅拌过夜。减压蒸除溶剂,残渣溶于乙酸乙酯,有机相以饱和食盐水洗,无水Na2SO4干燥,过滤,浓缩,残渣经反相HPLC分离,得白色固体(25mg,10.4%),即为最终所得糖原磷酸化酶抑制剂。产物鉴定数据如下:
ESI-MS m/z:1157.6[M+H]+;
1H NMR:(MeOD,400MHz)8.56(d,J=8.0Hz,1H),7.92-8.05(m,4H),7.65-7.74(m,2H),7.26-7.49(m,5H),7.15(d,J=6.0Hz,1H),6.95-7.04(m,2H),5.24-5.31(m,1H),4.80-4.86(m,1H),4.63(br s,1H),3.86(br s,1H),3.63-3.71(m,2H),3.35-3.56(m,9H),2.98-3.20(m,2H),268-2.75(m,2H),0.97(t,J=5.2Hz,3H),0.83(d,J=5.6Hz,3H),0.54(d,J=12.4Hz,3H)。
经鉴定产物具有式(I1)所示的结构式:
(1)本实施例所得式(I1)结构的产物用于体外GP酶活抑制活性测试:
1)显色剂:钼酸铵5g,500mL1M HCl,搅拌全部溶解后加入孔雀绿190mg,继续搅拌至全部溶解,锡纸避光保存;
2)缓冲液:a)精密称取Hepes 0.5958g,溶于5mL蒸馏水中,用10M的NaOH调pH至7.2,备用;b)精密称取氯化钾0.3728g,溶于5mL蒸馏水中,备用;c)精密称取氯化镁0.0255g,溶于1mL蒸馏水中,备用;d)精密称取EGTA 0.0476g,溶于5mL蒸馏水中,用10M的NaOH调pH至7.0,备用;e)精密称取G-1-P 0.0152g,溶于10mL蒸馏水中,备用;f)精密称取糖原10mg,溶于1mL蒸馏水中,备用。
3)阳性药溶液:精密称取一定量的咖啡因,溶于10mL蒸馏水中,配制成0.5、5、50、500μM的咖啡因溶液;
4)GPa溶液:取1μL的GPa加入到100μL的反应体系中,配制成终浓度为250ng/100μL的GPa溶液;
5)待测试化合物溶液:将待测试化合物溶于DMSO中配制成10mM的贮备液,取适量加入到反应体系中至不同终浓度;
6)实验步骤:(a)96孔板中设计PC(阳性对照)孔,Blank(空白对照)孔,阳性药孔(咖啡因)和待测化合物孔;(b)每孔加反应buffer 52μL;(c)在待测化合物孔加测试化合物至终浓度分别为1μM,5μM,10μM,25μM,50μM,100μM。(d)加酶1μL,终浓度为250ng/100μL。PC(阳性对照)孔只加酶,Blank(空白对照)孔不加酶;(e)加显色液150μL;(6)20-25℃条件下反应20min;(f)在波长655nm条件下比色;(g)结果的初步计算。其结果见表1:
抑制率=[(PC-B)-(X-B)]/(PC-B)。
表1.化合物的体外GP酶活抑制活性
(2)本实施例所得式(I1)结构的产物用于肝细胞糖原降解抑制活性测试:
分别采用大鼠肝细胞及人HepG2细胞,按照文献报道方法(Proc.Natl.Acas.Sci,1998,95,1776-1781;Brain Res.,2005,1060,89-99)测定化合物对糖原降解的抑制作用,结果见表2:
表2.化合物对肝细胞糖原分解的抑制作用
上述活性数据显示,本实施例式(I1)化合物具有糖原磷酸化酶的抑制作用,且其在细胞水平的抑制糖原分解作用与PSN-357相当。因此可用于制备抗糖尿病药物、抗脑缺血药物、抗心血管疾病药物、降血脂药物、减肥药物、抗动脉粥样硬化药物或抗肿瘤药物。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
一种糖原磷酸化酶抑制剂及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。









动态评分
0.0