

IPC分类号 : C07J63/00,A61K31/585,A61K31/56,A61P31/20






专利摘要
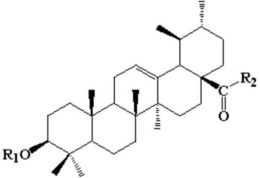
本发明公开了一种五环三萜类甘草次酸衍生物,具有以下式1所示通式或式7所示结构:各取代基定义详见说明书。本发明还公开了所述五环三萜类甘草次酸衍生物在制备抗HCV病毒的药物中的应用。Huh7细胞毒活性结果显示,甘草次酸衍生物,低细胞毒性,具有研究价值。体外抗HCVcc结果显示,甘草次酸抗HCVcc活性较弱,但其五环三萜类甘草次酸衍生物有较好抗HCV活性。
权利要求
1.一种五环三萜类甘草次酸衍生物,其特征在于,所述五环三萜类甘草次酸衍生物为以下的一种:
2.一种五环三萜类甘草次酸衍生物在制备抗HCV病毒的药物中的应用,其特征在于,所述五环三萜类甘草次酸衍生物为以下的一种:
3.根据权利要求2所述的五环三萜类甘草次酸衍生物在制备抗HCV病毒的药物中的应用,其特征在于,所述抗HCV病毒的药物是以所述五环三萜类甘草次酸衍生物为唯一的活性成份,或包含所述五环三萜类甘草次酸衍生物的药物组合物。
4.根据权利要求3所述的五环三萜类甘草次酸衍生物在制备抗HCV病毒的药物中的应用,其特征在于,所述包含五环三萜类甘草次酸衍生物的药物组合物是指所述五环三萜类甘草次酸衍生物与药学上允许的一种或多种辅料构成的药物组合物。
5.根据权利要求2所述的五环三萜类甘草次酸衍生物在制备抗HCV病毒的药物中的应用,其特征在于,所述五环三萜类甘草次酸衍生物可以和药剂学上的常规药用辅料制成药物制剂。
6.根据权利要求5所述的五环三萜类甘草次酸衍生物在制备抗HCV病毒的药物中的应用,其特征在于,所述药物制剂是胶囊剂、混悬剂、片剂、粉剂、乳剂、溶液剂、糖浆剂或注射剂中的至少一种。
说明书
技术领域
本发明属于医药技术领域,具体地说,涉及一种五环三萜类甘草次酸衍生物及制备方法与应用。
背景技术
丙型肝炎病毒(hepatitis C viral,HCV)是急、慢性肝炎的重要致病因子。慢性丙肝由于发病多,早期不易发现,诊断率低、反复发作和治疗难度大等特点,若没及时处理和干预,会逐渐发展为肝纤维化、肝硬化,甚至肝癌,从而成为威胁人类健康的重要疾病,备受关注。
甘草酸(glycyrrhizin,GL)具有治疗肝炎作用(Rossum v T G J.Glycyrrhizin treatment for Chronic Hepatitis C[J].Alimentary Pharmacology&Therapeutics,2000,12(3):199-205.),Matsumoto等研究报道甘草酸(glycyrrhizin,GL)可靶向于成熟的HCV病毒颗粒感染细胞后释放过程(Matsumoto Y,Matsuura T,Aoyagi H,et al.Antiviral activity of glycyrrhizin against hepatitis C virus in vitro.[J].Plos One,2013,8(7):e68992.)。18β-甘草次酸(18β-Glycyrrhetinic acid,GA)是一种来源于甘草(Glycyrrhiza glabra)的齐墩果烷型五环三萜酸,是甘草酸类药物代谢后起药理作用的主要物质(Kim D H,Lee S W,Han M J,et al.Biotransformation of glycyrrhizin to 18-beta-glycyrrhetinic acid-3-O-beta-D-glucuronide by Streptococcus LJ-22,a human intestinal bacterium[J].Biol Pharm Bull,1999,22(3):320-322.)。研究表明甘草次酸有保肝、抗炎、抗病毒(肝炎病毒、HIV、SARS等)及抗肿瘤等多种药理活性,临床上用于治疗消化道溃疡、乙型肝炎等疾病。在日本甘草酸类药物治疗慢性肝炎已有多年临床历史,其抗病毒效果已充分验证(Fujisawa K,Tandon B N.Therapeutic approach to the chronic active liver disease:Summary of a satellite symposium[J].Viral Hepatitis and Liver Disease,Springer,1994:662-665.)。日本学者报道,GL、GA和3-单葡萄糖醛酸甘草次酸有抗HBV活性,认为这3种药物通过抑制HBV感染细胞HBsAg分泌来达到抗HBV作用,其中GA活性最强,且无显著细胞毒性(Fujisawa K,Tandon B N.Therapeutic approach to the chronic active liver disease:Summary of a satellite symposium[J].Viral Hepatitis and Liver Disease,Springer,1994:662-665.蒲洁莹,何莉,吴思宇等,甘草属植物中三萜类化合物的抗病毒作用研究进展[J].病毒学报.2013,29(6):673-679.)。近来研究发现肝细胞表面有丰富的甘草次酸结合位点,因而甘草次酸及其衍生物能选择性地被肝细胞摄取,具有较好的肝靶向性,现已作为肝靶向药物载体使用(米雪,赵岩,杨慧.甘草次酸及其衍生物在肝靶向药物中的应用[J].天津药学。2014,26(1):41-45.)。
基于甘草次酸有治疗肝炎、抗病毒和肝靶向性等优点,结合前期课题组及周敏德课题组研究发现四环三萜酸和五环三萜酸均有较好的抑制HCV病毒入侵作用(Wang H,Wang Q,Xiao S L,et al.Elucidation of the pharmacophore of echinocystic acid,a new lead for blocking HCV entry[J].European Journal of Medicinal Chemistry,2013,64(6):160-168.Qian X J,Zhang X L,Ping Z,et al.A Schisandra-Derived Compound Schizandronic Acid Inhibits Entry of Pan-HCV Genotypes into Human Hepatocytes[J].Sci Rep,2016,6:27268.),本申请以甘草次酸为先导化合物,经氧化、成肟、重排及酯化、酰化设计合成一系列甘草次酸衍生物并进行抗HCVcc活性筛选,探究传统中药甘草中发挥抗肝炎物质,并初步探讨五环三萜类甘草次酸抗HCV构效关系。
发明内容
本发明的第一个目的是提供一种五环三萜类甘草次酸衍生物。
本发明的第二个目的是提供一种所述五环三萜类甘草次酸衍生物的制备方法。
本发明的第三个目的是提供一种所述五环三萜类甘草次酸衍生物在制备抗HCV病毒的药物中的应用。
为了实现上述目的,本发明采用的技术方案如下:
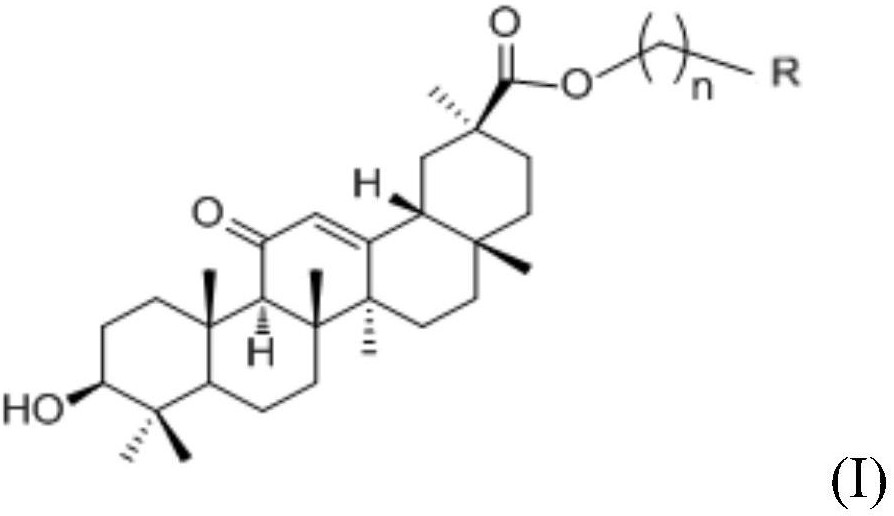
本发明的第一个方面提供了一种五环三萜类甘草次酸衍生物,具有以下式1所示通式或式7所示结构:
式1中,R1为羰基、肟基、羧酸基、氢、羟基、氰基、酯基、取代或未取代的烷基、取代或未取代的烷氧基、取代或未取代的芳基、取代或未取代的杂芳基;
R2为氢、羟基、氰基、取代或未取代的胺基、取代或未取代的烷基、取代或未取代的烷氧基、取代或未取代的芳基、取代或未取代的杂芳基、取代或未取代的环烷基。
较优选的,所述式1中,R1为羰基、肟基、羧酸基、氢、羟基、氰基、乙酯基、丙酯基、C1~10烷基、C1~10烷氧基、苯基、噻吩基、吡啶基、烷基取代苯基、卤代苯基、环己基、环丙基;
R2为氢、羟基、氰基、C1~10烷基、C1~10烷氧基、苯基、噻吩基、吡啶基、烷基取代苯基、卤代苯基、苯并三唑基、环己基、环丙基、
最优选的,所述五环三萜类甘草次酸衍生物为以下的一种:
本发明的第二个方面提供了一种所述五环三萜类甘草次酸衍生物的制备方法,包括以下步骤:
将摩尔比为(1.01~2):1(优选为1.15:1)的GA、PCC(氯铬酸吡啶)溶于二氯甲烷中,室温搅拌反应,柱层析提纯,得到化合物2;
将摩尔比为1:(2~4)(优选为1:3)的化合物2、NH2OH·HCl溶于无水吡啶中,室温搅拌反应,反应液用冰水淬灭,析出大量白色固体,抽滤,重结晶,得到化合物3;
将摩尔比为1:(2~4):(2~4)(优选为1:2:3)的化合物2、NaHCO3、m-CPBA溶于无水二氯甲烷中,室温搅拌反应,蒸除溶剂,继续放置过夜反应,柱层析提纯,得到化合物7;
将GA溶于吡啶中,滴加过量的酸酐,滴毕加热回流反应,反应液用冰水淬灭,抽滤,重结晶,得到化合物4;
所述酸酐为乙酸酐、丙酸酐。
将GA溶于醇中,滴加过量的浓盐酸或浓硫酸,加热回流反应,反应液用冰水淬灭,抽滤,重结晶,得到化合物5。
所述醇为甲醇、乙醇、正丙醇、1H-苯并三唑-1-甲醇。
将摩尔比为1:(2~4)的GA、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)溶于DIEA(N,N-二异丙基乙胺)中,滴加过量的有机胺,反应完全后用冰水淬灭,抽滤,重结晶,获得化合物6。
所述有机胺为甲胺、乙胺、正丙胺、正丁胺、环己胺、环丙基甲胺、环己甲胺、2-噻吩甲胺、4-氯苯甲胺。
本发明的第三个方面提供了一种所述五环三萜类甘草次酸衍生物在制备抗HCV病毒的药物中的应用。
所述抗HCV病毒的药物是以所述五环三萜类甘草次酸衍生物为唯一的活性成份,或包含所述五环三萜类甘草次酸衍生物的药物组合物。
所述包含五环三萜类甘草次酸衍生物的药物组合物是指所述五环三萜类甘草次酸衍生物与药学上允许的一种或多种辅料构成的药物组合物。
所述辅料为稀释剂、赋形剂、粘合剂、填充剂、崩裂剂、香味剂、甜味剂中的至少一种。
所述五环三萜类甘草次酸衍生物可以和药剂学上的常规药用辅料制成药物制剂。
所述药物制剂是胶囊剂、混悬剂、片剂、粉剂、乳剂、溶液剂、糖浆剂或注射剂中的至少一种。
所述药物制剂的给药方式为口服、注射。
由于采用上述技术方案,本发明具有以下优点和有益效果:
本申请是基于甘草次酸具有抗病毒作用和肝靶向性,结合四环三萜酸及五环三萜有较好的抑制HCV病毒入侵作用;合成一系列五环三萜类甘草次酸衍生物,利用HCVcc体外感染模型进行体外抗HCV活性筛选和对Huh7细胞毒活性测试。结果显示18个化合物均表现出较好的抗HCV活性,并且在细胞毒性方面,甘草次酸衍生物的细胞毒性都很低,具有深入开发前景。
附图说明
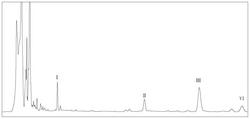
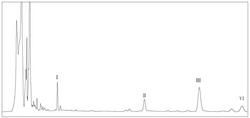
图1是本发明实施例制备的化合物抗HCVcc的IC50值柱状图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
本发明所使用的仪器:Broker Spectrospin AC-300P型核磁共振仪(德国布鲁克公司)、Synergy 2多功能酶标仪(美国BioTek公司)、CO2细胞培养箱(美国Thermo scientific公司)、CKX31倒置相差显微镜(日本Olympus公司)。
本发明所使用的试剂:化学试剂:18β-甘草次酸原料购于萨恩化学技术(上海)有限公司,其余分别购于阿拉丁试剂(上海)公司和安耐吉试剂公司,化学纯或分析纯;生物试剂:细胞活力检测试剂Cell Counting Kit-8(CCK-8)购自日本同仁公司。
实施例1
化合物2的合成方法如下:将GA(3000mg,6.40mmol)、PCC(氯铬酸吡啶)(1200mg,5.56mmol)溶于12mL二氯甲烷中,室温搅拌反应3h,蒸除溶剂,柱层析(石油醚:乙酸乙酯=1:1),得2550mg白色固体,产率85%,获得化合物2。MS:[M+H]+=469.76,1H-NMR(300MHz,CDCl3)δ5.75(s,1H,CH=),1.37(s,3H,Me-29),1.27(s,3H,Me-28),1.22(s,3H,Me-27),1.16(s,3H,Me-26),1.10(s,3H,Me-25),1.06(s,3H,Me-24),0.85(s,3H,Me-23).
化合物3的合成方法如下:将GA(240mg,0.51mmol)、NH2OH·HCl(106.32mg,1.53mmol)溶于4mL无水吡啶中,室温搅拌反应4h,反应液用100mL冰水淬灭,析出大量白色固体,抽滤,10mL 95%乙醇重结晶,得180mg白色固体,产率75%,获得化合物3。MS:[M+H]+=484.66,1H-NMR(300MHz,d-DMSO)δ12.20(d,J=3.4Hz,1H,-COOH),10.27(s,1H,-OH),5.41(s,1H,CH=),2.84(d,J=15.3Hz,1H),2.68–2.56(m,1H,H-12),1.34(s,3H,Me-29),1.13(s,3H,Me-28),1.09(d,J=3.0Hz,6H,Me-27、Me-26),1.06(s,3H,Me-25),0.98(s,3H,Me-24),0.75(s,3H,Me-23).
化合物7的合成方法如下:将化合物2(240mg,0.51mmol)、NaHCO3(s)(85.69mg,1.02mmol)、m-CPBA(264.03mg,1.53mmol)溶于4mL无水二氯甲烷中,室温搅拌反应15min,反应液蒸除溶剂,继续放置过夜反应,柱层析(石油醚:乙酸乙酯=2:1),得180mg白色固体,产率70%,获得化合物7。MS:[M-H]-=483.72,1H-NMR(300MHz,CDCl3)δ5.75(s,1H,CH=),1.47(s,3H,Me-29),1.37(s,3H,Me-28),1.21(s,6H,Me-27),1.14(s,3H,Me-26),1.10(s,3H,Me-25),1.06(s,3H,Me-24),0.83(s,3H,Me-23).
化合物4a合成:将GA(100mg,0.22mmol)溶于5mL吡啶中,滴加2.5mL乙酸酐,滴毕加热回流反应3h,反应液用100mL冰水淬灭,析出大量白色固体,抽滤,8mL 95%乙醇重结晶,得90mg白色固体,产率80%,获得化合物4a。MS:[M+H]+=513.21,1H-NMR(300MHz,CDCl3)δ5.65(s,1H,CH=),4.45(dd,J=11.4,4.8Hz,1H,H-3),1.99(s,3H,CH3),1.30(s,3H,Me-29),1.16(s,3H,Me-28),1.10(s,3H,Me-27),1.06(s,3H,Me-26),0.81(s,6H,Me-25、Me-24),0.77(s,3H,Me-23).
化合物4b合成:将GA(100mg,0.22mmol)溶于5mL吡啶中,滴加2.5mL丙酸酐,滴毕加热回流反应3h,反应液用100mL冰水淬灭,析出大量白色固体,抽滤,8mL 95%乙醇重结晶,得90mg白色固体,产率82%,获得化合物4b。MS:[M+H]+=526.89,1H-NMR(300MHz,CDCl3)δ5.28(s,1H),4.57–4.45(m,1H),2.33(q,J=7.5Hz,2H,CH2),1.14(d,J=4.5Hz,6H,CH3、Me-29),0.97–0.88(m,9H,Me-28、Me-27、Me-26),0.86(s,6H,Me-25、Me-24),0.75(s,3H,Me-23).
化合物5a合成:将GA(50mg,0.11mmol)溶于10mL甲醇中,滴加4mL浓盐酸,加热回流反应3.5h,反应液用100mL冰水淬灭,析出白色固体,抽滤,5mL 95%乙醇重结晶,得40mg白色固体,产率80%,获得化合物5a。MS:[M+H]+=484.66,1H-NMR(300MHz,CDCl3)δ5.64(s,1H,CH=),3.67(s,3H,CH3),3.21(dd,J=10.2,6.0Hz,1H,H-3),1.34(s,3H,Me-29),1.13(s,3H,Me-28),1.11(s,3H,Me-27),1.10(s,3H,Me-26),0.98(s,3H,Me-25),0.79(s,6H,Me-24、Me-23).
化合物5b合成:将GA(50mg,0.11mmol)溶于10mL乙醇中,滴加4mL浓盐酸,加热回流反应3.5h,反应液用100mL冰水淬灭,析出白色固体,抽滤,5mL 95%乙醇重结晶,得43.6mg白色固体,产率85%,获得化合物5b。MS:[M+H]+=499.93,1H-NMR(300MHz,CDCl3)δ5.58(s,1H,CH=),4.14(q,J=7.1Hz,2H,CH2),3.23(dd,J=10.9,5.3Hz,1H,H-3),1.35(s,3H,Me-29),1.28(d,J=7.1Hz,3H,Me-28),1.22(s,3H,Me-27),1.20(s,3H,Me-26),1.14(s,3H,Me-25),1.01(s,3H,Me-24),0.81(s,3H,Me-23),0.72(s,3H,CH3).
化合物5c合成:将GA(50mg,0.11mmol)溶于10mL正丙醇中,滴加4mL浓盐酸,加热回流反应3.5h,反应液用100mL冰水淬灭,析出白色固体,抽滤,5mL 95%乙醇重结晶,得42.3mg白色固体,产率80%,获得化合物5c。MS:[M+H]+=513.78,1H-NMR(300MHz,CDCl3)δ5.55(s,1H,CH=),4.03(t,J=6.6Hz,2H,CH2),3.21(dd,J=10.8,5.2Hz,1H,H-3),1.33(s,3H,Me-29),1.21(s,3H,Me-28),1.18(s,3H,Me-27),1.12(s,3H,Me-26),0.98(s,3H,Me-25),0.79(s,6H,Me-24 Me-23),0.70(s,3H,CH3).
化合物5d合成:将GA(50mg,0.11mmol)溶于10mL 1H-苯并三唑-1-甲醇中,滴加4mL浓盐酸,加热回流反应3.5h,反应液用100mL冰水淬灭,析出白色固体,抽滤,5mL 95%乙醇重结晶,得44.6mg白色固体,产率90%,获得化合物5d。MS:[M+H]+=588.68,1H-NMR(300MHz,CDCl3)δ8.09(d,J=8.4Hz,1H,ArH),7.57(t,J=7.6Hz,1H,ArH),7.44(t,J=7.6Hz,1H,ArH),7.35(d,J=8.3Hz,1H,ArH),5.71(s,1H,CH=),3.23(dd,J=9.8,6.1Hz,1H,H-3),1.58(s,3H,Me-29),1.41(s,3H,Me-27),1.16(s,3H,Me-28),1.13(s,3H,Me-26),1.01(s,3H,Me-25),0.94(s,3H,Me-24),0.81(s,3H,Me-23).
化合物6a合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)中,滴加2mL甲胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL 95%乙醇重结晶得190mg白色固体,产率85%,获得化合物6a。MS:[M+H]+=484.92,1H-NMR(300MHz,CDCl3)δ5.60(s,1H,CH=),3.23–3.09(m,1H,3-H),2.76(d,J=4.7Hz,3H,CH3),1.30(s,3H,Me-29),1.06(s,9H,Me-28、Me-27、Me-26),0.94(s,3H,Me-25),0.74(d,J=2.7Hz,6H,Me-24、Me-23).
化合物6b合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)出中,滴加2mL乙胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL95%乙醇重结晶得190mg白色固体,产率82%,获得化合物6b。MS:[M+H]+=498.91,1H-NMR(300MHz,CDCl3)δ5.65(s,2H,CH=,OH),3.34(dt,J=12.7,6.0Hz,2H,CH2),3.25(d,J=6.5Hz,1H,3-CH),1.38(d,J=4.6Hz,6H,Me-29、Me-27),1.13(d,J=5.6Hz,9H,Me-28、Me-26、Me-25),1.01(s,3H,Me-24),0.81(d,J=2.1Hz,6H,CH3、Me-23).
化合物6c合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)中,滴加2mL正丙胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL 95%乙醇重结晶得190mg白色固体,产率83%,获得化合物6c。MS:[M+H]+=512.85,1H-NMR(300MHz,CDCl3)δ5.66(s,1H,CH=),5.63(d,1H,-OH),3.24(dd,J=12.4,6.7Hz,3H,H-3,CH2),1.40(s,3H,Me-29),1.38(s,3H,Me-27),1.14(s,6H,Me-28、Me-26),1.02(s,3H,Me-25),0.94(t,J=7.4Hz,3H,Me-24),0.82(s,3H,Me-23),0.82(s,3H,CH3).
化合物6d合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)中,滴加2mL正丁胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL 95%乙醇重结晶得190mg白色固体,产率86%,获得化合物6d。MS:[M+H]+=527.15,1H-NMR(300MHz,CDCl3)δ5.56(s,1H,CH=),5.49(d,1H,-OH),4.04(dd,J=14.2,7.1Hz,1H,H-3),3.28–3.09(m,2H,CH2),1.29(s,3H,Me-29),1.04(s,9H,Me-28、Me-27、Me-26),0.92(s,3H,Me-25),0.85(t,J=7.2Hz,3H,CH3),0.73(s,6H,Me-24、Me-23).
化合物6e合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)中,滴加2mL环己胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL 95%乙醇重结晶得190mg白色固体,产率85%,获得化合物6e。MS:[M+H]+=552.91,1H-NMR(300MHz,CDCl3)δ5.63(s,1H,CH=),5.49(d,J=8.3Hz,1H,-OH),3.88–3.71(m,1H,CH),3.23(dd,J=10.2,6.0Hz,H-3,1.37(s,3H,Me-29),1.14(s,3H,Me-28),1.13(s,3H,Me-27),1.12(s,3H,Me-26),1.01(s,3H,Me-25),0.81(s,6H,Me-24、Me-23).
化合物6f合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)中,滴加2mL环丙基甲胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL 95%乙醇重结晶得190mg白色固体,产率84.8%,获得化合物6f。MS:[M+H]+=524.89,1H-NMR(300MHz,CDCl3)δ5.58(s,1H,CH=),3.22–3.08(m,2H,CH2),3.07–2.95(m,1H,H-3),1.31(s,3H,Me-29),1.06(s,9H,Me-28、Me-27、Me-26),0.94(s,3H,Me-25),0.74(d,J=2.9Hz,6H,Me-24、Me-23),0.49–0.39(m,2H,CH2),0.14(q,J=4.9Hz,2H,CH2).
化合物6g合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)中,滴加2mL环己甲胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL 95%乙醇重结晶得190mg白色固体,产率80%,获得化合物6g。MS:[M+H]+=567.11,1H-NMR(300MHz,CDCl3)δ5.68(d,J=5.7Hz,1H,-OH),5.66(s,1H,CH=),3.24(dd,J=10.1,6.2Hz,1H,H-3),3.12(dt,J=13.8,6.9Hz,2H,CH2),1.40(s,3H,Me-29),1.38(s,3H,Me-28),1.14(s,6H,Me-27、Me-26),1.01(s,3H,Me-25),0.82(d,J=1.2Hz,6H,Me-24、Me-23).
化合物6h合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)中,滴加2mL 2-噻吩甲胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL 95%乙醇重结晶得190mg白色固体,产率83%,获得化合物6h。MS:[M+H]+=566.21,1H-NMR(300MHz,CDCl3)δ7.15(d,J=4.5Hz,1H,CH),6.88(d,J=4.2Hz,2H,2×CH),5.99(s,1H,OH),5.52(s,1H,CH=),4.57(qd,J=15.2,5.5Hz,2H,CH2),3.20–3.08(m,1H,H-3),1.29(s,3H,Me-29),1.07(s,3H,Me-28),1.05(s,6H,Me-27、Me-26),0.93(s,3H,Me-25),0.73(s,6H,Me-24、Me-23).
化合物6i合成:将GA(200mg,0.43mmol)、PyBOP(六氟磷酸苯并三唑-1-基-氧基三吡咯烷)(244.6mg,1.47mmol)溶于2mL DIEA(N,N-二异丙基乙胺)中,滴加2mL 4-氯苯甲胺,加毕室温继续搅拌反应4h,反应液用100mL冰水淬灭,析出白色固体,抽滤,10mL 95%乙醇重结晶得190mg白色固体,产率80%,获得化合物6i。MS:[M+H]+=595.76(这个是不是差的有点多),1H-NMR(300MHz,CDCl3)δ7.34(dt,J=14.8,7.4Hz,1H,Ar-H),6.89–6.76(m,2H,2×Ar-H),6.15(s,1H,-OH),5.58(s,1H,CH=),4.50–4.43(m,2H,CH2),3.32–3.16(m,1H,H-3),1.36(s,3H,Me-29),1.12(s,9H,Me-28、Me-27、Me-26),1.00(s,3H,Me-25),0.81(s,3H,Me-24),0.77(s,3H,Me-23).
实施例2
本发明实施例1合成的五环三萜类甘草次酸衍生物的体外抗HCVcc活性测试
一.实验药物、试剂及材料
所用试剂购自Sigma公司。
1.细胞系Huh7,人肝癌细胞株(详见:Yimin Tong,Yongzhe Zhu,Xueshan Xia,Yuan Liu,et al.Tupaia CD81,SR-BI,Claudin-1,and Occludin Support Hepatitis C Virus Infection,JOURNAL OF VIROLOGY,2011;85(6):2793-2802;Jin Zhong,Pablo Gastaminza,Guofeng Cheng,et al.Robust hepatitis C virus infection in vitro,PNAS,2005;102(26):9294-9299)。
2.细胞培养液配制,含10%胎牛血清、0.03%谷氨酰胺、非必需氨基酸、氨苄青霉素和链霉素100U/ml,调pH至7.4。
3.细胞消化液配制,含0.25%胰蛋白酶,用磷酸缓冲液配制。
4.HCVcc:细胞培养的感染性丙型肝炎病毒(详见:Yimin Tong,Yongzhe Zhu,Xueshan Xia,Yuan Liu,et al.Tupaia CD81,SR-BI,Claudin-1,and Occludin Support Hepatitis C Virus Infection,JOURNAL OF VIROLOGY,Mar.2011;85(6):2793-2802;Jin Zhong,Pablo Gastaminza,Guofeng Cheng,et al.Robust hepatitis C virus infection in vitro,PNAS,2005;102(26):9294-9299)。
二、实验方法
(一)HCVcc的制备
1.病毒扩增
HCVcc病毒(丙肝病毒)来源:日本重症肝炎1型(Japanese fulminant hepatitis type 1,JFH-1)基因的质粒是由日本东京国立传染性研究所的Wakita友情提供,并用来表达得到JFH-1HCVcc。
JFH-1嵌合HCVcc(105ffu/ml),取50μl感染接种于24孔板的Huh7.5细胞,次日换液,随后根据细胞生长密度传代培养,观察细胞生长状态,待病毒快速增殖导致的细胞病变效应(cytopathic effect,CPE)出现后,收集第7-20天的培养上清,取0.1ml用于病毒滴度测定,其余8000rpm离心5min弃细胞碎片后分装保存于-70℃备用。
2.病毒滴定
以空白Huh7.5(1×104cells/孔)、12h,弃培养上清,每孔加入100μl经10倍梯度稀释的HCVcc上清液,共孵育5h,换新鲜DMEM全培养液继续培养72h,行免疫荧光法检测HCV阳性细胞,一抗用1:100稀释的HCV抗体阳性病人血清,二抗为1:100稀释的FITC标记羊抗人IgG。在荧光显微镜下观察发光细胞,并记录最后一个可观察到绿色荧光阳性细胞的孔内绿色荧光阳性细胞数及相应的稀释梯度,计算出focus forming unit/ml(ffu/ml)数值,以此代表HCVcc滴度。
(二)HCVcc感染性的检测
取处于对数生长期的Huh7细胞,调整细胞浓度为1×105个/ml,取100μl种96孔板;培养24h后加入待测化合物及HCVcc,化合物按0、10、25、50、100μg/ml浓度稀释,37℃培养4h后去除化合物及HCVcc混液,换培养基继续培养;48h后进行免疫荧光检测,在荧光显微镜下读取各孔HCVcc阳性克隆数。本发明实施例制备的化合物体外抗HCVcc活性结果如表1和图1所示:大部分表现出有较好的抗HCV活性,除化合物4a、4b、6b、6c和7之外均显现较好的活性,其中2、5a、5c、6e共4个衍生物活性均优于甘五酸。
表1目标化合物抗HCV活性结果
本发明是基于甘草次酸具有治疗肝炎、抗病毒和肝靶向性等特点,设计、合成一系列甘草次酸类衍生物并研究其抗HCV活性,对于探究传统中药甘草中发挥抗肝炎物质、初步探讨甘草次酸衍生物抗HCV构效关系及丰富五环三萜类化合物抗HCV研究,具有意义。
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
一种五环三萜类甘草次酸衍生物及制备方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0