

IPC分类号 : C07J63/00I,A61K31/58I,A61P31/16I






专利摘要
本发明公开了含氮杂环甘草次酸衍生物及其制备方法和抗甲型流感病毒应用。本发明的甘草次酸衍生物包含氮杂环,本发明合成了一系列具有抗IAV活性的甘草次酸衍生物。而且本发明的甘草次酸衍生物对甲型流感病毒(IAV)具有较强的抑制活性,明显强于常用的阳性药利巴韦林具对甲型流感病毒的抑制性,说明本发明的甘草次酸衍生物能够用于制备抗甲型流感病毒(IAV)药物的应用且没有副作用。
权利要求
1.具有抗IAV活性的甘草次酸衍生物,其特征在于结构式如下所示:
2.权利要求1所述甘草次酸衍生物的用途,其特征在于所述甘草次酸衍生物用于制备抗甲型流感病毒(IAV)药物的应用。
3.一种甘草次酸衍生物的制备方法,其特征在于步骤如下:
其中,阿拉伯数字Ⅰ-1~Ⅲ-13代表反应的化合物,
甘草次酸分别与1,2-二溴乙烷和1,3-二溴丙烷发生取代反应,得到化合物1和2,化合物1分别与咪唑和哌嗪发生S
化合物1、2、Ⅰ-1~Ⅰ-4的结构式如下所示:
其中,R
另外,
甘草次酸与乙酸酐反应,得到3-乙酰基甘草次酸,即化合物3,化合物3分别与1,2-二溴乙烷和1,3-二溴丙烷反应,得到化合物4和化合物5;化合物4与哌嗪和1,2,4-三氮唑反应,得到化合物Ⅰ-5和Ⅰ-6;化合物5与咪唑反应,得到化合物Ⅰ-7;
化合物3~5、Ⅰ-5~Ⅰ-7的结构式如下所示:
其中,R
另外,
甘草次酸与氯铬酸吡啶盐反应,得到3-氧代甘草次酸,即化合物6,化合物6与亚硝酸异戊酯反应,生成3-氧代甘草次酸-2-肟,即化合物7,化合物7与盐酸羟胺反应,生成甘草次酸-2,3-二肟,即化合物8,化合物8与氢氧化钠和次氯酸钠反应,得到化合物9,化合物9分别与草酰氯和1,2-二溴乙烷反应,分别得到化合物10和化合物11;化合物10与哌嗪反应,得到化合物Ⅱ-8,化合物Ⅱ-8与氯乙腈反应得到化合物Ⅱ-9;化合物11分别与咪唑和哌嗪反应,得到化合物Ⅱ-10和化合物Ⅱ-11;
化合物6~11、Ⅱ-8~Ⅱ-11的结构式如下所示:
另外,
化合物6与三溴化吡啶鎓反应,生成化合物2-溴-3-氧代甘草次酸,即化合物12,化合物12与硫脲反应,生成化合物13;化合物13与1,2-二溴乙烷反应,生成化合物14;化合物14分别与咪唑和哌嗪反应,分别生成化合物Ⅲ-12和化合物Ⅲ-13;化合物Ⅲ-13与氯乙腈反应,生成化合物Ⅲ-14;
化合物12~14、Ⅲ-12~Ⅲ-14的结构式如下所示:
说明书
技术领域
本发明涉及医药技术领域,具体的说,是新型甘草次酸衍生物、甘草次酸衍生物的制备方法及其在抗甲型流感病毒领域的用途。
背景技术
甲型流感病毒(influenza A virus,IAV)是每年季节性流感的主要病原体,也是全球急性呼吸道感染的重要病毒性病原,且其耐药性出现迅速。因此,研究开发新结构类型抗IAV化合物,对更好地抵御流感病毒对人类健康的侵害方面具有重要意义。
甘草次酸(glycyrrhetinic acid,GA),又称beta-甘草亭酸、甘珀酸,是甘草的主要活性成分之一,由甘草酸水解脱除两分子的葡萄糖醛酸所得的苷元,属于齐墩果烷型骨架的五环三萜烯类化合物。
甘草次酸的化学结构如下:
研究表明,GA具有抗炎、抗菌、抗肿瘤、抗氧化、抗利尿、抗病毒及抗糖尿病等多种药理作用,已被广泛应用于食品、烟草、医药和化妆品等行业。但是长期给予高剂量的GA通常会引起假性醛固酮过多症,导致严重的高血压和心脏肥大,限制了GA的实际应用。因此,研究人员以GA为先导物进行了大量的化学结构修饰,以期获得生物活性较好及细胞毒性较低的化合物。
发明内容
为解决现有技术中的问题,本发明目的在于提供一种具有更好抗IAV活性的甘草次酸衍生物及其制备方法和应用。本发明的甘草次酸衍生物具有抗IAV活性且细胞毒性低,能够用于制备抗IAV药物的应用。
本发明具有抗IAV活性的甘草次酸衍生物,其结构通式为Ⅰ、Ⅱ、Ⅲ所示:
其中,R1、R2、R3和R4如表1、表2和表3所示:
表1通式Ⅰ所代表的化合物Ⅰ-1~Ⅰ-7
表2通式Ⅱ所代表的化合物Ⅱ-8~Ⅱ-11
表3通式Ⅲ所代表的化合物Ⅲ-12~Ⅲ-14
本发明的另一目的在于提供通式为Ⅰ、Ⅱ、Ⅲ的甘草次酸衍生物的制备方法。
本发明的通式为Ⅰ、Ⅱ、Ⅲ的甘草次酸衍生物的制备方法,包括下列步骤:
其中,阿拉伯数字1~14代表反应的化合物,如1即为化合物1,具体结构式见表4~表6。
(1)甘草次酸分别与1,2-二溴乙烷和1,3-二溴丁烷发生取代反应,得到化合物1和化合物2,化合物1与咪唑和哌嗪发生SN2取代反应,得到化合物Ⅰ-1和化合物Ⅰ-2;化合物2与咪唑和1,2,4-三氮唑反应得到化合物Ⅰ-3和化合物Ⅰ-4。
所述化合物Ⅰ-1~Ⅰ-4制备方法的反应路线如下:
试剂和条件:(a)DMF,K2CO3,1,2-dibromoethane/1,3-dibromopropane,1.5h,r.t.;(b)DMF,K2CO3,imidazole/piperazine/1,2,4-triazole,r.t.
(2)甘草次酸与乙酸酐反应得到3-乙酰基甘草次酸(3),化合物3分别与1,2-二溴乙烷和1,3-二溴丁烷反应,得到化合物4和化合物5;化合物4与哌嗪和1,2,4-三氮唑反应得到化合物Ⅰ-5和Ⅰ-6;化合物5与咪唑反应,得到化合物Ⅰ-7。
所述化合物Ⅰ-5~Ⅰ-7制备方法的反应路线如下:
试剂和条件:(c)Ac2O,pyridine,r.t.;(a)DMF,K2CO3,1,2-dibromoethane/1,3-dibromopropane,1.5h,r.t.;(b)DMF,K2CO3,imidazole/piperazine/1,2,4-triazole,r.t.
(3)甘草次酸与氯铬酸吡啶盐反应,得到3-氧代甘草次酸(6),化合物6与亚硝酸异戊酯反应,生成3-氧代甘草次酸-2-肟(7),化合物7与盐酸羟胺反应,生成甘草次酸-2,3-二肟(8),化合物8与氢氧化钠和次氯酸钠反应,得到化合物9。化合物9与草酰氯和1,2-二溴乙烷反应,分别得到化合物10和化合物11;化合物10与哌嗪反应,得到化合物Ⅱ-8,化合物Ⅱ-8与氯乙腈反应,得到化合物Ⅱ-9;化合物11与咪唑和哌嗪反应,得到化合物Ⅱ-10和Ⅱ-11。
所述化合物Ⅱ-8~Ⅱ-11制备方法的反应路线如下:
试剂和条件:(d)PCC,DCM,r.t.;(e)isoamyl nitrite,t-BuOK,t-BuOH,r.t.;(f)hydroxylamine hydrochloride,pyridine,r.t.;(g)EtOH,NaOH,NaOCl,r.t.;(h)DCM,oxalyl chloride,-5℃,1h,2h,r.t.;(i)DCM,piperazine,triethylamine,3.5h,r.t.;(j)DMF,chloroacetonitrile,K2CO3,r.t.;(a)DMF,K2CO3,1,2-dibromoethane,1.5h,r.t.;(b)DMF,K2CO3,imidazole/piperazine,r.t..
(4)化合物6与三溴化吡啶鎓反应,生成化合物2-溴-3-氧代甘草次酸(12),化合物12与硫脲反应,生成化合物13;化合物13与1,2-二溴乙烷反应,生成化合物14;化合物14分别与咪唑哌嗪反应,生成化合物Ⅲ-12和化合物Ⅲ-13;化合物Ⅲ-13与氯乙腈反应,生成化合物Ⅲ-14。
所述化合物Ⅲ-12~Ⅲ-14制备方法的反应路线如下:
试剂和条件:(d)PCC,DCM,r.t.;(k)pyridiniumtribromide,AcOH,r.t.;(l)thiocarbamide,ethanol,80℃,4h;(m)1,2-dibromoethane,DMF,K2CO3,r.t.;(n)DMF,K2CO3,imidazole/piperazine,r.t.;(j)DMF,chloroacetonitrile,K2CO3,r.t.
其中化合物1~7为合成通式Ⅰ系列目标化合物过程的中间产物,如下所示,
其中,它们的相关基团R1、R2如表4所示:
表4通式Ⅰ系列化合物的中间体1~7
其中,化合物7和化合物8为通式Ⅱ系列化合物合成过程中的中间产物,结构如下所示:
化合物9~11为通式Ⅱ系列目标化合物合成过程中的中间产物,如下所示:
其中,基团R3如表5所示:
表5通式Ⅱ系列化合物的中间体9~11
化合物12为通式Ⅲ系列化合物合成过程中的中间产物,结构如下所示:
化合物13和化合物14为通式Ⅲ系列目标化合物合成过程中的中间产物,如表6所示。
表6通式Ⅲ系列化合物的中间体13和14
本发明的另一目的在于提供通式为Ⅰ、Ⅱ、Ⅲ的甘草次酸衍生物的用途。
通式为Ⅰ、Ⅱ、Ⅲ的甘草次酸衍生物用于制备抗甲型流感病毒(IAV)药物的用途。
药理试验表明本发明的甘草次酸衍生物对甲型流感病毒(IAV)具有较强的抑制活性,明显强于常用的阳性药利巴韦林具对甲型流感病毒的抑制性,而且,部分化合物如I-2和I-7同时具有较低的细胞毒性,说明本发明的甘草次酸衍生物能够用于制备抗甲型流感病毒(IAV)药物的应用且没有副作用。
本发明的优点在于获得通式I、Ⅱ、Ⅲ的一系列甘草次酸新衍生物。而且,药理学实验结果表明本发明的化合物I-2、I-6、I-7、II-10、II-11、III-12和III-13相比于阳性对照药利巴韦林具对甲型流感病毒具有更好的抑制活性;尤其是化合物I-2和I-7同时具有较低的细胞毒性,作为成药应用时没有副作用,具有进一步临床开发应用的价值;以及,化合物II-11和III-13对甲型流感病毒的抑制活性远强于阳性药利巴韦林,可以作为药物先导分子进一步研究开发,提高其成药性。
附图说明
图1为化合物I-2浓度变化对甲型流感病毒的抑制率。
图2为化合物I-7浓度变化对甲型流感病毒的抑制率。
图3为10uM浓度的甘草次酸衍生物与利巴韦林的抗IAV病毒结果。
具体实施方式
按照上述内容,在不脱离本发明上述基本技术思想的前提下,根据本领域的普通技术知识和惯用手段,对其内容还可以有多种形式的修改、替换或变更。下面通过具体实施例,对本发明的实施内容再进一步详细说明,但它们不是对本发明的限定。凡是基于本发明上述内容实现的技术均属于本发明的范围。
下面结合具体实施例对本发明作进一步的说明。
实施例1
化合物Ⅰ-1的合成
向20mL圆底烧瓶中加入5mL DMF,加入GA(200.0mg,0.425mmol,1.0eq),室温搅拌,原料未全部溶解,再依次加入1,2-二溴乙烷(638.0mg,3.4mmol,8.0eq)和K2CO3(117.0mg,0.85mmol,2.0eq)。TLC(乙酸乙酯/石油醚=1/3,v/v)监控反应进程,1.5h原料消失,停止反应。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,迅速析出大量白色沉淀,抽滤,用适量水洗,滤饼自然晾干,得到白色固体,即化合物1,216.0mg,收率88.0%。直接用于后续反应。
向10mL反应瓶中加入5mLDMF,室温搅拌,加入化合物1(200.1mg,0.346mmol,1.0eq),原料部分溶解,再依次加入咪唑(90.0mg,1.321mmol,4.0eq)和K2CO3(143.2mg,1.038mmol,3.0eq)。TLC(乙酸乙酯/甲醇=10/1,v/v)监控反应进程,3h原料消失,停止反应。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,迅速析出大量白色沉淀,抽滤,水洗,自然晾干后得到粗品,经硅胶柱层析,采用梯度洗脱(乙酸乙酯/石油醚=7:1~4:1,v/v),得到142.7mg浅黄色固体,为目标化合物Ⅰ-1,收率73.1%。m.p.248.1~250.3℃。1H NMR(400MHz,CDCl3)δ7.49(s,1H),7.07(s,1H),6.94(s,1H),5.59(s,1H),4.34(dd,J=11.7,5.7Hz,2H),4.21(t,J=5.4Hz,2H),3.70(q,J=7.0Hz,1H),3.21(dd,J=10.7,5.6Hz,1H),2.85-2.67(m,1H),2.31(s,1H),1.97(d,J=4.2Hz,1H),1.83-1.77(m,1H),1.62(d,J=3.4Hz,1H),1.58(s,2H),1.33(s,3H),1.22(t,J=7.0Hz,1H),1.11(s,3H),1.09(s,3H),1.07(s,3H),0.78(s,3H),0.75(s,3H).HR-MS:calcd for C35H52N2O4[M+H]+565.3999,found565.3996.
实施例2
化合物Ⅰ-2的合成
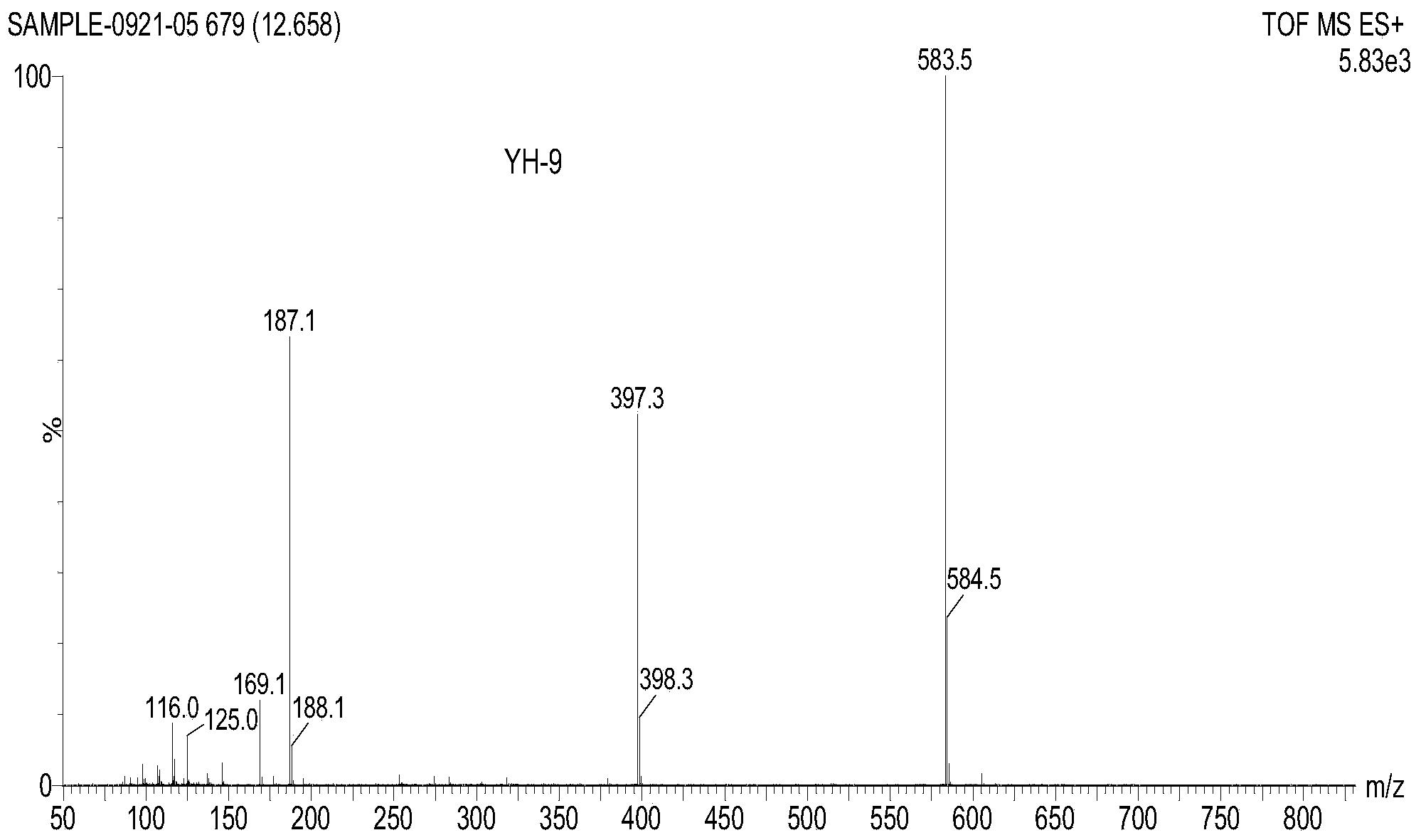
向10mL反应瓶中加入5mLDMF,室温搅拌,加入化合物1(200.1mg,0.346mmol,1.0eq),原料部分溶解,再依次加入哌嗪(113.8mg,1.320mmol,4.0eq)和K2CO3(143.2mg,1.038mmol,3.0eq)。TLC(乙酸乙酯/甲醇=10/1,v/v)监控反应进程,3h原料消失,停止反应。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,迅速析出大量白色沉淀,抽滤,水洗,自然晾干后得到粗品,经硅胶柱层析,采用梯度洗脱(乙酸乙酯/石油醚=7:1~4:1,v/v),得到138.0mg类白色固体,为目标化合物Ⅰ-2,收率68.4%,m.p.235.2~238.7℃。1H NMR(400MHz,CH3OD)δ5.62(s,1H),4.33(dd,J=11.0,6.5Hz,1H),4.16(dd,J=12.0,6.1Hz,1H),3.58(q,J=7.1Hz,1H),3.29-3.28(m,1H),3.21(d,J=4.9Hz,2H),2.74(dd,J=8.5,3.8Hz,3H),2.44(s,1H),2.12(dd,J=18.5,9.2Hz,2H),1.94(d,J=11.3Hz,1H),1.80-1.70(m,1H),1.40(s,3H),1.14(s,3H),1.11(s,3H),0.97(s,3H),0.81(s,3H),0.77(s,3H).13CNMR(100MHz,CH3OD)δ201.32,176.61,171.63,127.51,78.00,61.85,60.59,56.46,54.80,49.62,48.65,45.45,43.97,43.56,43.27,41.01,38.96,38.90,37.54,36.98,34.78,33.46,32.41,31.64,30.69,27.99,27.31,27.24,26.46,26.23,25.99,22.51,17.93,17.24,15.62,14.98.HR-MS:calcd for C36H58N2O4[M+H]+583.4469,found 583.4467.
实施例3
化合物Ⅰ-5的合成
向50mL反应瓶中加入25mL吡啶和GA(5.00g,10.5mmol,1.0eq),室温搅拌,再加入乙酸酐(2.14g,21.0mmol,2.0eq)和4-二甲氨基吡啶(125.0mg,1.05mmol,0.1eq),TLC(乙酸乙酯/石油醚=1/1,v/v)监控反应进程。反应2h,原料消失,停止反应。开启磁力搅拌,将反应液缓慢倾入100mL冰水中,再加入适量稀盐酸,析出白色沉淀,抽滤,水洗,自然晾干,得到白色固体,即化合物3,5.28g,收率98.2%。
向20mL反应瓶中加入5mLDMF和化合物3(250.0mg,0.488mmol,1.0eq),室温搅拌,原料部分溶解,再依次加入1,2-二溴乙烷(733.2mg,3.904mmol,8.0eq)和K2CO3(135.4mg,0.976mmol,2.0eq),TLC(石油醚/乙酸乙酯=4/1,v/v)监控反应进程,1.5h反应完全。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,析出大量白色沉淀,抽滤,水洗,滤饼自然晾干,得白色固体,即化合物4,239.7mg,收率79.0%。直接用于后续反应。
向20mL反应瓶中加入5mLDMF和化合物4(200.2mg,0.322mmol,1.0eq),室温搅拌,再依次加入哌嗪(113.9mg,1.322mmol,4.0eq)和K2CO3(134.5mg,0.966mmol,3.0eq),TLC(乙酸乙酯/甲醇=10/1,v/v)监控反应进程,3h反应结束。后处理过程参照化合物Ⅰ-1的合成,粗品经硅胶柱分离纯化,得到目标化合物Ⅰ-5,浅黄色固体,收率73.2%,m.p.198.6~204.1℃。1H NMR(400MHz,CH3OD)δ5.61(s,1H),4.46(dd,J=11.8,4.6Hz,1H),4.40-4.24(m,1H),4.24-4.05(m,1H),3.58(q,J=7.0Hz,1H),3.28(dt,J=3.3,1.6Hz,4H),3.05(dd,J=4.9,2.3Hz,4H),2.81-2.53(t,4H),2.48(s,1H),2.20-2.08(m,2H),2.01(s,3H),1.88(s,1H),1.74(t,J=13.3Hz,3H),1.41(s,4H),1.26(s,1H),1.14(d,J=2.0Hz,6H),0.88(d,J=3.6Hz,9H),0.82(s,3H).13C NMR(101MHz,CH3OD)δ201.01,176.61,171.55,171.53,127.54,100.00,80.83,61.66,60.78,56.99,56.75,54.77,51.25,45.43,44.07,43.94,43.30,41.01,38.41,37.72,37.56,36.90,32.31,31.65,30.70,27.95,27.25,27.16,26.24,26.01,23.14,22.53,19.80,17.94,17.11,17.03,15.80,15.64.HR-MS:calcd forC38H61O5N2[M+H]+625.4575,found 625.4558.
实施例4
化合物Ⅰ-7的合成
向20mL反应瓶中加入5mLDMF和化合物3(250.0mg,0.488mmol,1.0eq),室温搅拌,原料部分溶解,再依次加入1,3-二溴丙烷(787.5mg,3.901mmol,8.0eq)和K2CO3(135.4mg,0.976mmol,2.0eq),TLC(石油醚/乙酸乙酯=4/1,v/v)监控反应进程,1.5h反应完全。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,析出大量白色沉淀,抽滤,水洗,滤饼自然晾干,得白色固体,即化合物5,244.1mg,收率79.0%。直接用于后续反应。
向20mL反应瓶中加入5mLDMF和化合物5(200.0mg,0.316mmol,1.0eq),室温搅拌,再依次加入咪唑(85.9mg,1.262mmol,4.0eq)和K2CO3(130.9mg,0.947mmol,3.0eq),TLC(乙酸乙酯/甲醇=10/1,v/v)监控反应进程,3h反应结束。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,迅速析出大量白色沉淀,抽滤,水洗,自然晾干后得到粗品,经硅胶柱层析,采用梯度洗脱(乙酸乙酯/石油醚=7:1~4:1,v/v),得到目标化合物Ⅰ-7,类白色固体,收率73.8%,m.p.206.3~209.5℃。1H NMR(400MHz,CDCl3)δ7.60(s,1H),7.10(s,1H),6.94(s,1H),5.62(s,1H),4.50(dd,J=11.6,4.8Hz,1H),4.11-4.00(m,8H),3.03-2.55(m,1H),2.35(s,3H),2.04(s,8H),1.64-1.59(m,4H),1.36(s,3H),1.24(s,3H),1.15(d,J=5.7Hz,3H),1.11(s,3H),0.87(s,3H),0.80(s,3H).13C NMR(100MHz,CDCl3)δ200.02,176.20,171.08,169.09,137.17,129.95,128.61,118.84,80.66,61.83,60.94,55.09,48.51,45.48,44.14,43.76,43.30,41.10,38.87,38.12,37.02,37.87,32.77,31.95,30.49,28.67,28.48,28.12,26.50,26.44,23.63,23.44,21.38,18.76,16.75,16.48.HR-MS:calcd forC38H57O5N2[M+H]+621.4262,found 621.4280.
实施例5
化合物Ⅱ-10的合成
以DCM为溶剂,PCC为氧化剂,合成化合物3-氧代甘草次酸(6),得到白色固体,收率80.2%。
向100mL反应瓶中加入30mL叔丁醇,室温搅拌,加入化合物6(1.01g,2.1mmol,1.0eq),原料全部溶解,再依次加入叔丁醇钾(1.18g,10.5mmol,5.0eq)、亚硝酸异戊酯(0.74g,6.3mmol,3.0eq),TLC(石油醚/乙酸乙酯=3/1,v/v)监控反应进程,2h原料消失,停止反应。减压除去叔丁醇,粗品用150mLDCM溶解,缓慢滴加稀盐酸调节pH=7,加入50mL水萃取,分液,有机相再用水、饱和食盐水洗涤3遍,干燥,过滤,减压浓缩,经柱层析纯化(石油醚/乙酸乙酯=5/1~3/1,v/v),得3-氧代甘草次酸-2-肟(7)810.3mg,收率76.0%。
向10mL反应瓶中加入5mL吡啶,室温搅拌,加入化合物7(800.3mg,1.61mmol,1.0eq),原料全部溶解,再加入盐酸羟胺(280.5mg,4.03mmol,2.5eq),110℃油浴加热,TLC(二氯甲烷/甲醇=10/1,v/v)监控反应进程,2.5h原料消失,停止反应。待反应液降至室温,边搅拌边滴加冰水,逐渐析出淡粉红色沉淀,抽滤,滤饼用60mL乙酸乙酯溶解,滴加稀盐酸调pH至中性,再用水、饱和氯化钠溶液洗涤两次,干燥,过滤,减压浓缩,得到甘草次酸-2,3-二肟(8)775.9mg,收率94.1%。
向50mL反应瓶中加入25mL无水乙醇,室温下开启磁力搅拌,加入化合物8(500.2mg,0.98mmol,1.0eq),原料全部溶解,再依次加入氢氧化钠(100.0mg,2.48mmol,2.5eq),2mL氯酸钠溶液,TLC(二氯甲烷/甲醇=15/1,v/v)监控反应,0.5h原料消失。减压浓缩,剩余物用50mL乙酸乙酯溶解,再依次用水、饱和氯化钠水溶液洗涤,干燥,过滤,减压浓缩得到粗品,经柱层析纯化(石油醚/乙酸乙酯=5/1,v/v)得到化合物9,白色固体,295.2mg,收率59.0%。
向20mL反应瓶中加入5mLDMF和化合物9(250.0mg,0.490mmol,1.0eq),室温搅拌,原料部分溶解,再依次加入1,2-二溴乙烷(735.7mg,3.916mmol,8.0eq)和K2CO3(135.4mg,0.976mmol,2.0eq),TLC(石油醚/乙酸乙酯=4/1,v/v)监控反应进程,1.5h反应完全。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,析出大量白色沉淀,抽滤,水洗,滤饼自然晾干,得白色固体,即化合物11,219.8mg,收率72.7%。直接用于后续反应。
向20mL反应瓶中加入5mL DMF和化合物11(200.0mg,0.324mmol,1.0eq),室温搅拌,再依次加入咪唑(88.2mg,1.295mmol,4.0eq)和K2CO3(134.3mg,0.972mmol,3.0eq),TLC(乙酸乙酯/甲醇=10/1,v/v)监控反应进程,3h反应结束。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,迅速析出大量白色沉淀,抽滤,水洗,自然晾干后得到粗品,经硅胶柱层析,采用梯度洗脱(乙酸乙酯/石油醚=7:1~4:1,v/v),得到化合物Ⅱ-10,浅黄色固体,收率56.5%,m.p.238.5~243.3℃。1H NMR(400MHz,CDCl3)δ7.49(s,1H),7.07(s,1H),6.95(s,1H),5.68(s,1H),4.47-4.28(m,2H),4.22(t,J=5.3Hz,2H),3.95(d,J=17.3Hz,1H),3.69(dd,J=7.0,0.7Hz,1H),2.51(s,1H),2.01(d,J=18.0Hz,3H),1.67(dd,J=10.8,7.7Hz,1H),1.51(dd,J=15.5,3.8Hz,2H),1.41(s,3H),1.35(s,8H),1.15(s,3H),1.11(s,3H),1.08(s,3H),0.78(s,3H).13C NMR(101MHz,CDCl3)δ198.19,175.98,170.00,163.22,130.05,128.45,111.50,63.09,59.57,52.41,48.25,45.85,45.24,44.12,43.46,41.13,37.95,37.90,37.70,35.49,34.60,31.91,31.89,31.49,31.04,30.73,29.39,28.59,26.57,26.35,23.96,23.38,18.52,18.13,16.45.HR-MS:calcd for C35H49O5N4[M+H]+605.3698,found 605.3680.
实施例6
化合物Ⅱ-11的合成
向20mL反应瓶中加入5mL DMF和化合物11(200.0mg,0.324mmol,1.0eq),室温搅拌,再依次加入哌嗪(111.6mg,1.295mmol,4.0eq)和K2CO3(134.3mg,0.972mmol,3.0eq),TLC(乙酸乙酯/甲醇=10/1,v/v)监控反应进程,3h反应结束。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,迅速析出大量白色沉淀,抽滤,水洗,自然晾干后得到粗品,经硅胶柱层析,采用梯度洗脱(乙酸乙酯/石油醚=7:1~4:1,v/v),得到化合物Ⅱ-11,浅黄色固体,收率63.7%,m.p.242.1~244.7℃。1H NMR(400MHz,CH3OD)δ5.73(s,1H),4.39-4.28(m,1H),4.25-4.11(m,1H),3.74(d,J=17.1Hz,1H),3.58(q,J=7.0Hz,1H),3.38-3.20(m,3H),3.12(s,3H),2.69(d,J=4.9Hz,6H),2.18(dd,J=23.6,10.3Hz,2H),1.88(s,1H),1.43(d,J=8.2Hz,6H),1.35(s,3H),1.15(s,3H),1.10(s,3H),0.84(s,3H).HR-MS:calcd forC36H55O5N4[M+H]+623.4167,found 623.4156.
实施例7
化合物Ⅲ-12的合成
向500mL反应瓶中加入150mL冰醋酸,室温下开启磁力搅拌,加入化合物6(3.00g,6.41mmol,1.0eq),原料全部溶解,再加入三溴化吡啶(2.25g,7.05mmol,1.1eq),TLC(石油醚/乙酸乙酯=2/1,v/v)监控反应进程,1h原料消失,停止反应。减压蒸干,剩余物用适量乙酸乙酯溶解,分别用饱和碳酸钠水溶液和饱和氯化钠溶液洗涤,干燥,过滤,减压浓缩,得到黄色固体3.21g,即化合物12,收率92.0%。未经纯化,直接用于下一步反应。
向500mL反应瓶中加入200mL无水乙醇,室温磁力搅拌,加入化合物12(3.87g,7.09mmol,1.0eq),再加入硫脲(2.05g,28.30mmol,4.0eq),置于80℃油浴加热。TLC(二氯甲烷/甲醇=10/1,v/v)监控反应进程,4h原料消失,停止反应。减压蒸干,剩余物用乙酸乙酯溶解,分别用水、饱和食盐水洗涤有机相,减压浓缩,经柱层析纯化,得到化合物13,浅黄色固体,2.67g。
向20mL反应瓶中加入5mL DMF和化合物13(250.0mg,0.488mmol,1.0eq),室温搅拌,原料部分溶解,再依次加入1,2-二溴乙烷(716.0mg,3.811mmol,8.0eq)和K2CO3(134.9mg,0.976mmol,2.0eq),TLC(石油醚/乙酸乙酯=4/1,v/v)监控反应进程,1.5h反应完全。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,析出大量白色沉淀,抽滤,水洗,滤饼自然晾干,得浅黄色固体,即化合物14,223.9mg,收率74.4%。直接用于后续反应。
向20mL反应瓶中加入5mL DMF和化合物14(200.0mg,0.317mmol,1.0eq),室温搅拌,再依次加入咪唑(86.2mg,1.266mmol,4.0eq)和K2CO3(131.4mg,0.951mmol,3.0eq),TLC(乙酸乙酯/甲醇=10/1,v/v)监控反应进程,3h反应结束。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,迅速析出大量白色沉淀,抽滤,水洗,自然晾干后得到粗品,经硅胶柱层析,采用梯度洗脱(乙酸乙酯/石油醚=7:1~4:1,v/v),得到粉红色固体,即化合物Ⅲ-12,108.3mg,收率55.2%,m.p.235.5~237.9℃。1H NMR(400MHz,CH3OD)δ7.80(s,1H),7.22(s,1H),7.04(s,1H),5.56(s,1H),4.36(s,3H),3.79-3.45(m,3H),3.28(dt,J=3.5,1.6Hz,4H),3.16-3.01(m,1H),2.61(s,1H),2.20(d,J=16.0Hz,1H),1.76(d,J=4.8Hz,3H),1.39(s,3H),1.26(s,3H),1.19(s,3H),1.13(d,J=5.2Hz,6H),1.06(s,3H),0.77(s,3H).13C NMR(100MHz,CH3OD)δ186.26,176.18,171.65,167.65,148.95,137.39,127.70,127.34,119.64,114.27,89.67,87.19,79.65,75.83,72.64,62.99,60.03,56.99,52.23,43.95,43.38,40.96,37.94,36.28,31.61,29.15,27.77,27.15,26.28,25.99,22.44,22.38,21.19,18.48,17.47,15.07.HR-MS:calcd for C36H51O3N4S[M+H]+619.3676,found619.3655.
实施例8
化合物Ⅲ-13的合成
向20mL反应瓶中加入5mL DMF和化合物14(200.0mg,0.317mmol,1.0eq),室温搅拌,再依次加入哌嗪(109.2mg,1.268mmol,4.0eq)和K2CO3(131.4mg,0.951mmol,3.0eq),TLC(乙酸乙酯/甲醇=10/1,v/v)监控反应进程,3h反应结束。开启磁力搅拌,将反应液缓慢倾入50mL冰水中,迅速析出大量白色沉淀,抽滤,水洗,自然晾干后得到粗品,经硅胶柱层析,采用梯度洗脱(乙酸乙酯/石油醚=7:1~4:1,v/v),得到粉红色固体,即化合物Ⅲ-13,127.8mg,收率63.3%,m.p.240.7~242.9℃。1H NMR(400MHz,CDCl3)δ7.34(dq,J=8.2,2.9Hz,1H),7.13(ddd,J=11.0,7.5,5.3Hz,1H),6.56(s,1H),5.56(s,1H),5.18-4.99(m,1H),2.85(s,1H),2.46(dt,J=3.7,1.8Hz,6H),1.87(s,1H),1.36(d,J=4.4Hz,1H),1.20(dd,J=8.7,4.5Hz,3H),1.10(s,3H),1.05(s,3H),1.02(s,1H),1.01-0.98(m,1H),0.83(s,3H).13C NMR(100MHz,CDCl3)δ199.81,199.36,176.38,170.16,128.59,60.39,59.27,52.53,45.35,45.08,44.10,43.84,43.42,42.60,38.16,37.98,36.90,36.85,35.68,31.93,30.46,30.43,28.81,28.48,23.42,22.47,22.46,22.42,20.90,20.86,18.35,16.19,16.13,15.99.HR-MS:calcd.for C37H57O3N4S[M+H]+637.4146,found 637.4126.
实施例9
合成的甘草次酸新衍生物的抗甲型流感病毒(IAV)活性测试
稳定表达Gluc报告系统的293T细胞系的建立
将重组质粒pLenti6-Gluc质粒转染293T细胞,在Blasticidin选择性压力下连续传代。经过抗性筛选存活下来的细胞克隆中,胞内Ⅰ型RNA聚合酶将会转录出一种vRNA样的负链RNA。该RNA两端分别为流感病毒基因组节段上的3'UTR和5'UTR,中间是反义的Gluc编码序列。一旦病毒感染细胞,流感病毒聚合酶可识别这种负链RNA,把其当做病毒vRNA,经转录产生带有Gluc编码序列的mRNA,继而合成Gluc蛋白。
经过筛选,从获得的一系列可稳定表达Gluc报告系统的293T细胞系中,挑选效果最好的克隆6,命名其为293T-Gluc用于后续实验。该细胞在流感病毒感染时,合成Gluc报告蛋白并分泌到培养基上清中,通过检测荧光素酶的活性即可实现对病毒感染性的相对定量。
293T-Gluc模型对化合物抗流感活性的评价
将293T-Gluc细胞培养至90%贴壁,用0.25%胰蛋白酶消化后,以7×105细胞/毫升的浓度在含10%FBS的DMEM中培养,配制成一定浓度的细胞悬液。然后将细胞接种于96孔板,每孔加入细胞悬液100μL,置5%CO2、37℃培养24h。预先加入药物孵育2h,然后将病毒原毒稀释以MOI=0.3接种病毒。于37℃继续培养24h,测定感染细胞中荧光素酶活性。在每个96孔板中,利巴韦林用作阳性对照。下式计算测试化合物的抑制率,
计算出抑制率后,利用统计分析软件绘制细胞生长抑制率曲线,通过曲线拟合得到函数计算抑制率在50%时对应的化合物浓度即为其对该细胞的半数抑制浓度(IC50)。
甘草次酸衍生物抗IAV病毒活性结果见表7。
表7甘草次酸衍生物抗IAV病毒结果
*NA=No Activity;NT=Not tested
需要说明的是,本发明选取利巴韦林作阳性药对照药,为已知的流感病毒抑制剂。
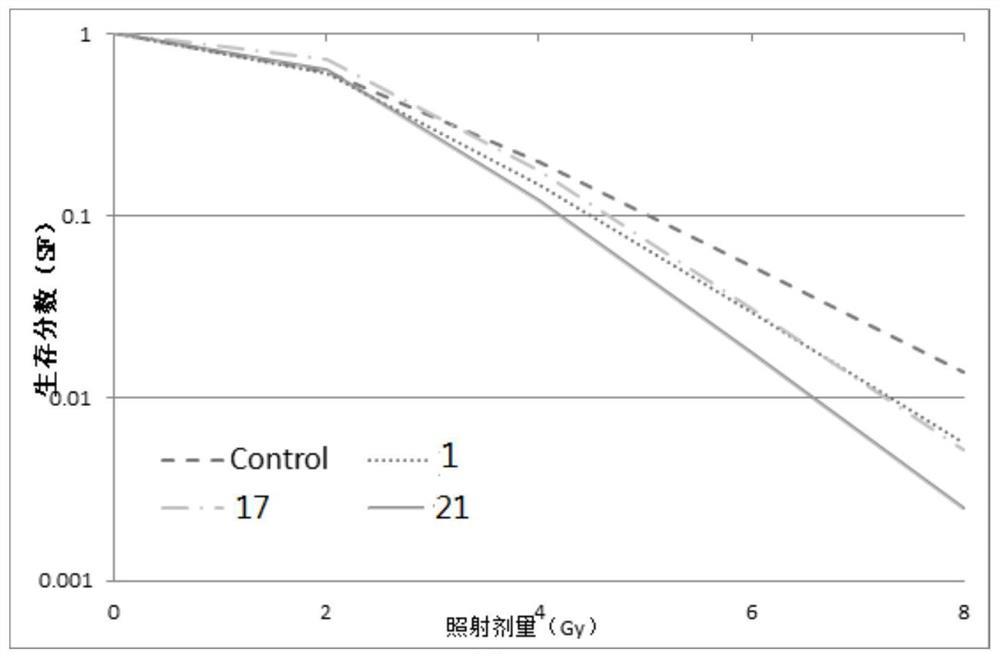
本发明通过甘草次酸20位引入哌嗪侧链得到的酯类衍生物对甲型流感病毒的抑制活性较强,而且,基于目前获得的药理实验数据表明,本发明的甘草次酸2、3位骈合氧化呋咱杂环后其酯类衍生物对甲型流感病毒的抑制活性明显优于酰胺类衍生物。即药理学实验结果证明:如图3所示,本发明的甘草次酸新衍生物对甲型流感病毒表现出明显的抑制性,尤其是化合物I-2、I-6、I-7、II-10、II-11、III-12和III-13在10μM浓度下对甲型流感病毒的抑制率分别为相同浓度阳性药利巴韦林的2.8、2.6、2.6、3.1、3.7、2.5和3.8倍,说明本发明的化合物相比于阳性对照药利巴韦林具对甲型流感病毒具有更好的抑制作用,例如,如图1和图2所示,表明化合物I-2、I-7在浓度为20uM时即可对甲型流感病毒达到比较稳定的抑制效果;而且,同等药物浓度条件下本发明化合物Ⅰ-2、Ⅰ-7的CC50分别为76.93±4.32μM和>100μM,表明本发明的化合物I-2和I-7的具有较低的细胞毒性,作为成药应用时具有较低的副作用,具有进一步临床开发应用的价值;再者,化合物II-11和III-13对甲型流感病毒的抑制活性远强于阳性药利巴韦林,可以作为药物先导分子进一步研究开发,提高其成药性。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种等同变换,这些等同变换均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
含氮杂环甘草次酸衍生物及其制备方法和抗甲型流感病毒应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












动态评分
0.0