








专利摘要
本发明涉及蓝色电致发光高分子材料及其制备方法。本发明基于深蓝色高分子可以向蓝光或蓝绿光荧光染料分子发生有效能量转移的物理思想,以聚芴为深蓝色高分子母体,以具有高荧光量子效率的蓝光或蓝绿光萘酰亚胺单元为掺杂客体单元,将掺杂客体单元化学接枝到深蓝色高分子母体上,实现掺杂客体单元在深蓝色高分子母体中的分子水平分散,通过调节蓝光或蓝绿光荧光染料分子在深蓝色高分子母体的相对含量,实现深蓝色高分子向蓝光或蓝绿光荧光染料分子的部分或完全能量转移,增强蓝光或蓝绿光荧光染料分子发光,继而实现高分子材料的纯蓝色电致发光,构造出一类分子分散型蓝色电致发光高分子材料。
权利要求
1.一种蓝色电致发光高分子聚芴类材料,其特征在于具有如下结构:
其中:R1为己基、辛基、碳原子数为1-10的烷基取代的苯基、4-(二苯基胺基)苯基;R2为氢或碳原子数为1-10的烷基;
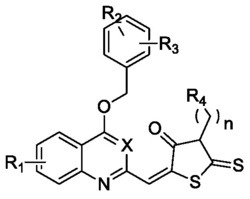
Ar为萘酰亚胺衍生物基元,具有如下一种或两种以下结构单元:
第(I)类:4位二烷基氨基取代的萘酰亚胺基元
其中,R3为氢或碳原子数为1-10的烷基,R4为氢、碳原子数为1-10的烷基或烷氧基或二烷基氨基、硝基、腈基、氨基、二苯基胺基;
第(II)类:4位二芳基氨基取代的萘酰亚胺基元
其中,X为氧原子或硫原子;
第(III)类:4位碳原子取代的萘酰亚胺基元
其中,1≤p≤5;
第(IV)类:4位其他杂原子取代的萘酰亚胺基元
x为萘酰亚胺基元含量,满足0<x≤1,m=1-20,n=1-300。
2.一种制备权利要求1所述蓝色电致发光高分子材料的方法,涉及三种单体和一种共聚合反应:
(1).2,7-双溴代芴及其衍生物单体的制备
2,7-双溴代芴及其衍生物单体的结构通式为:
2,7-双溴代芴及其衍生物单体包括两类:9,9-二烷基-2,7-双溴代芴单体和9,9-二芳基-2,7-双溴代芴单体,其制备方法分别如下:
1)9,9-二烷基-2,7-双溴代芴单体的制备
将2,7-二溴代芴和过量的溴代烷烃溶于甲苯中,再加入氢氧化钠水溶液,在氮气保护下反应,将反应产物倒入水中,分离有机相,反复水洗数次后,干燥、浓缩,重结晶,获得9,9-二烷基-2,7-二溴代芴;
2)9,9-二芳基-2,7-双溴代芴单体的制备
首先,将2,7-二溴代芴酮溶于乙醚中,在氮气保护下加入芳基格氏试剂,回流反应,制备出9-羟基-9-芳基-2,7-二溴代芴,将9-羟基-9-芳基-2,7-二溴代芴缓慢滴到芳烃的硫酸溶液中,回流反应,将反应产物倒入水中,分离有机相,反复水洗数次后,干燥、浓缩,重结晶,获得9,9-二芳基-2,7-二溴代芴;
(2).2,7-双硼酸酯芴及其衍生物单体的制备
2,7-双硼酸酯芴及其衍生物单体的结构通式为:
2,7-双硼酸酯芴及其衍生物单体的制备方法如下:
将正丁基锂在-78-0℃下加入到9,9-二取代-2,7-二溴代芴的四氢呋喃溶液中,搅拌反应,在-78-0℃下加入过量的硼酸三甲酯,室温搅拌反应,将反应混合物倒入水中,分离有机相,反复水洗数次后,干燥、浓缩,得到的产物用甲苯溶解,再加入摩尔比为2-3倍的1,3-丙二醇,回流,将反应产物倒入水中,分离有机相,反复水洗数次后,干燥、浓缩,乙醇重结晶,获得2,7-双硼酸酯芴及其衍生物;
(3).双溴代萘酰亚胺衍生物单体的制备
双溴代萘酰亚胺衍生物单体主要包括双溴代芴单萘酰亚胺衍生物单体和双溴代芴双萘酰亚胺衍生物单体;
1)溴代芴单萘酰亚胺衍生物单体
其中Ar为萘酰亚胺衍生物基元;
双溴代芴单萘酰亚胺衍生物单体的制备方法如下:
首先,将9-取代-2,7-二溴代芴和摩尔比为1-6倍的链节数为m的双溴代烷烃溶于甲苯中,再加入摩尔比为1-50倍的10-70%的氢氧化钠水溶液和摩尔比为0.005-0.2的四丁基溴化铵,在氮气保护下30-100℃反应1-24小时,制备出如上结构的9-取代-9-(m-溴烷基)-2,7-二溴代芴;然后,将萘酰亚胺衍生物Ar2溶于DMSO中,加入摩尔比为1-10倍的氢氧化钾,然后加入摩尔比为1-5倍的9-取代-9-(m-溴烷基)-2,7-二溴代芴,在20-150℃温度下反应1-50小时后,用水终止反应,分离有机相,反复水洗数次后,干燥、浓缩,柱分离,获得双溴代芴单萘酰亚胺衍生物单体;
2)双溴代芴双萘酰亚胺衍生物单体
其中Ar为萘酰亚胺衍生物基元;
双溴代芴双萘酰亚胺衍生物单体的制备方法如下:
首先,将2,7-二溴代芴和摩尔比为2-6倍的链节数为m的双溴代烷烃溶于甲苯中,再加入摩尔比为2-100倍的10-70%的氢氧化钠水溶液和摩尔比为0.005-0.2的四丁基溴化铵,在氮气保护下30-100℃反应1-72小时,制备出如上结构的9,9-(m-溴烷基)-2,7-二溴代芴;然后,将萘酰亚胺衍生物Ar1溶于DMSO中,加入摩尔比为1-10倍的氢氧化钠,然后加入摩尔比为0.1-1倍的9,9-(m-溴烷基)-2,7-二溴代芴,在20-150℃温度下反应1-50小时后,用水终止反应,分离有机相,反复水洗数次后,干燥、浓缩,柱分离,获得双溴代芴双萘酰亚胺衍生物单体;
(4)蓝色电致发光高分子材料的制备
蓝色电致发光高分子材料的制备采用Suzuki聚合反应,其制备方法如下:
将2,7-双硼酸酯芴衍生物单体、0.001%-100%摩尔比的双溴代萘酰亚胺衍生物单体、0-100%摩尔比的2,7-双溴代芴衍生物单体、0-100%摩尔比的电子传输单元或空穴传输单元单体溶解在甲苯中,然后加入摩尔比为2-10倍的2.0M碳酸钾水溶液;在氮气保护和50-100℃温度下,加入0.05%-10%摩尔比的四(三苯基膦)合钯,0-50%摩尔比的Aliquat 336;反应24-120小时后,经氯仿萃取、水洗、干燥、浓缩、甲醇沉降、溶剂抽提、真空干燥,最后获得纤维状高分子材料。
说明书
技术领域技术领域
本发明涉及蓝色电致发光高分子聚芴类材料及其制备方法。
技术背景技术背景
自1990年英国剑桥大学Cavendish实验室首次报导了共轭聚合物的电致发光二极管以来,高分子电致发光材料与器件由于具有工艺简单、成本低廉、易于实现大屏幕显示和柔性显示等突出特点受到学术界和产业界的广泛关注和竞相投入。为了实现高分子发光的全色显示,人们需要开发能够发射红绿蓝三基色的电致发光高分子材料。目前,红光和绿光高分子材料已经达到了实用化的要求,而蓝光高分子材料的各项指标,包括效率、寿命,离实用化尚有较大的差距,因此,成为制约高分子发光显示屏产业化的主要瓶颈之一。
聚芴及其衍生物是典型的蓝光高分子,具有很高的荧光量子效率,很好的光、热和化学稳定性,被认为是最具有应用潜力的高分子蓝光材料之一。为了提高聚芴及其衍生物器件的电致发光效率,通常采取在聚合物的主链或端基上引入载流子传输单元,调节电致发光器件的载流子平衡,达到提高其电致发光效率的目的。例如:美国DowChemical公司通过将具有空穴传输性能的三芳胺单元共聚到聚芴的主链中,获得了电致发光效率为2.82cd/A的蓝光聚芴类高分子材料(Microelectronics Journal,35,343-348,2004)。德国U.Scherf研究小组用三芳胺单元对聚芴进行封端以后,得到的高分子发光材料,其电致发光效率可达1.1cd/A(Advanced Materials13,565-570,2001)。
发明内容发明内容
本发明的目的是提供一类分子分散型蓝色电致发光高分子材料。
本发明的另一目的是提供一种蓝色电致发光高分子材料的制备方法。
本发明基于深蓝色高分子可以向蓝光或蓝绿光荧光染料分子发生有效能量转移的物理思想,以聚芴为深蓝色高分子母体,以具有高荧光量子效率的蓝光或蓝绿光萘酰亚胺单元为掺杂客体单元,将掺杂客体单元化学接枝到深蓝色高分子母体上,实现掺杂客体单元在深蓝色高分子母体中的分子水平分散。通过调节蓝光或蓝绿光荧光染料分子在深蓝色高分子母体的相对含量,实现深蓝色高分子向蓝光或蓝绿光荧光染料分子的部分或完全能量转移,增强蓝光或蓝绿光荧光染料分子发光,继而实现高分子材料的纯蓝色电致发光,构造出一类分子分散型蓝色电致发光高分子材料。
本发明所提供的分子分散型蓝色电致发光高分子材料具有如下结构:
其中:R1为己基、辛基、碳原子数为1-10的烷基取代的苯基、4-(二苯基胺基)苯基;R2为氢或碳原子数为1-10的烷基;
Ar为萘酰亚胺衍生物基元,具有如下一种或两种结构单元:
第(I)类:4位二烷基氨基取代的萘酰亚胺基元
其中,R3为氢或碳原子数为1-10的烷基,R4为氢、碳原子数为1-10的烷基或烷氧基或二烷基氨基、硝基、腈基、氨基、二苯基胺基。
第(II)类:4位二芳基氨基取代的萘酰亚胺基元
其中,X为氧原子或硫原子。
第(III)类:4位碳原子取代的萘酰亚胺基元
其中,1≤p≤5。
第(IV)类:4位其他杂原子取代的萘酰亚胺基元
x为萘酰亚胺基元含量,满足0<x≤1,m=1-20,n=1-300。
本发明提供的蓝色电致发光高分子材料的制备方法主要涉及三种单体和一种共聚合反应:
(1).2,7-双溴代芴及其衍生物单体的制备
2,7-双溴代芴及其衍生物单体的结构通式为:
2,7-双溴代芴及其衍生物单体包括两类:9,9-二烷基-2,7-双溴代芴单体和9,9-二芳基-2,7-双溴代芴单体,其制备方法分别如下:
1)9,9-二烷基-2,7-双溴代芴单体的制备
将2,7-二溴代芴和过量的溴代烷烃溶于甲苯中,再加入氢氧化钠水溶液,在氮气保护下反应,将反应产物倒入水中,分离有机相,反复水洗数次后,干燥、浓缩,重结晶,获得9,9-二烷基-2,7-二溴代芴;
2)9,9-二芳基-2,7-双溴代芴单体的制备
首先,将2,7-二溴代芴酮溶于乙醚中,在氮气保护下加入芳基格氏试剂,回流反应,制备出9-羟基-9-芳基-2,7-二溴代芴,将9-羟基-9-芳基-2,7-二溴代芴缓慢滴到芳烃的硫酸溶液中,回流反应,将反应产物倒入水中,分离有机相,反复水洗数次后,干燥、浓缩,重结晶,获得9,9-二芳基-2,7-二溴代芴。
(2).2,7-双硼酸酯芴及其衍生物单体的制备
2,7-双硼酸酯芴及其衍生物单体的结构通式为:
2,7-双硼酸酯芴及其衍生物单体的制备方法如下:
将正丁基锂在-78-0℃下加入到9,9-二取代-2,7-二溴代芴的四氢呋喃溶液中,搅拌反应,在-78-0℃下加入过量的硼酸三甲酯,室温搅拌反应,将反应混合物倒入水中,分离有机相,反复水洗数次后,干燥、浓缩,得到的产物用甲苯溶解,再加入摩尔比为2-3倍的1,3-丙二醇,回流,将反应产物倒入水中,分离有机相,反复水洗数次后,干燥、浓缩,乙醇重结晶,获得2,7-双硼酸酯芴及其衍生物。
(3).双溴代萘酰亚胺衍生物单体的制备
双溴代萘酰亚胺衍生物单体主要包括双溴代芴单萘酰亚胺衍生物单体和双溴代芴双萘酰亚胺衍生物单体。
1)双溴代芴单萘酰亚胺衍生物单体
其中Ar为萘酰亚胺衍生物基元;
双溴代芴单萘酰亚胺衍生物单体的制备方法如下:
首先,将9-取代-2,7-二溴代芴和摩尔比为1-6倍的链节数为m的双溴代烷烃溶于甲苯中,再加入摩尔比为1-50倍的10-70%的氢氧化钠水溶液和摩尔比为0.005-0.2的四丁基溴化铵,在氮气保护下30-100℃反应1-24小时,制备出如上结构的9-取代-9-(m-溴烷基)-2,7-二溴代芴。然后,将萘酰亚胺衍生物溶于DMSO中,加入摩尔比为1-10倍的氢氧化钾,然后加入摩尔比为1-5倍的9-取代-9-(m-溴烷基)-2,7-二溴代芴,在20-150℃温度下反应1-50小时后,用水终止反应,分离有机相,反复水洗数次后,干燥、浓缩,柱分离,获得双溴代芴单萘酰亚胺衍生物单体。
2)双溴代芴双萘酰亚胺衍生物单体
其中Ar为萘酰亚胺衍生物基元;
双溴代芴双萘酰亚胺衍生物单体的制备方法如下:
首先,将2,7-二溴代芴和摩尔比为2-6倍的链节数为m的双溴代烷烃溶于甲苯中,再加入摩尔比为2-100倍的10-70%的氢氧化钠水溶液和摩尔比为0.005-0.2的四丁基溴化铵,在氮气保护下30-100℃反应1-72小时,制备出如上结构的9,9-(m-溴烷基)-2,7-二溴代芴。然后,将萘酰亚胺衍生物溶于DMSO中,加入摩尔比为1-10倍的氢氧化钠,然后加入摩尔比为0.1-1倍的9,9-(m-溴烷基)-2,7-二溴代芴,在20-150℃温度下反应1-50小时后,用水终止反应,分离有机相,反复水洗数次后,干燥、浓缩,柱分离,获得双溴代芴双萘酰亚胺衍生物单体。
(4)蓝色电致发光高分子材料的制备
蓝色电致发光高分子材料的制备采用Suzuki聚合反应,其制备方法如下:
将2,7-双硼酸酯芴衍生物单体、0.001%-100%摩尔比的双溴代萘酰亚胺衍生物单体、0-100%摩尔比的2,7-双溴代芴衍生物单体、0-100%摩尔比的电子传输单元或空穴传输单元单体溶解在甲苯中,然后加入摩尔比为2-10倍的2.0M碳酸钾水溶液。在氮气保护和50-100℃温度下,加入0.05%-10%摩尔比的四(三苯基膦)合钯,0-50%摩尔比的Aliquat 336。反应24-120小时后,经氯仿萃取、水洗、干燥、浓缩,甲醇沉降、溶剂抽提、真空干燥,最后获得纤维状高分子材料。
附图说明说明书附图
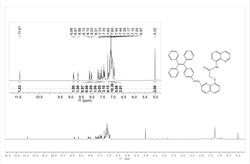
图1是蓝色电致发光高分子的固态吸收光谱和荧光发射光谱。
图2是基于蓝色电致发光高分子的单层器件,结构为ITO/PEDOT/蓝色电致发光高分子/Ca/Al的电致发光光谱。
从图1中可以看出,该材料的最大吸收波长在392nm;其荧光发射光谱有两个峰,分别在427nm和474nm;从图2中可以看出,该材料的电致发光光谱的峰值在472nm,并且423nm还有一个肩峰;
具体实施方式具体实施方式
实施例1:2,7-二溴-9-癸基-9-(2-(4-氨基-1,8-萘酰亚胺-9-)乙基-1-)芴
在氮气气氛保护下,将2.12g(10.0mmol)4-胺基-1,8-萘酰亚胺溶于60毫升二甲基亚砜,再向溶液中加入0.84g(15.0mmol)粉末化的氢氧化钾,电磁搅拌下于50℃反应十分钟,再向体系中逐渐加入3.426g(6.0mmol)2,7-二溴-9-癸基-9-(2-溴乙基-1-)芴。反应14小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9-癸基-9-(2-(4-氨基-1,8-萘酰亚胺-9-)乙基-1-)芴2.99g,产率71%。
实施例2:2,7-二溴-9-癸基-9-(2-(4-二甲基氨基-1,8-萘酰亚胺-9-)乙基-1-)芴
在氮气气氛保护下,将0.351g(0.5mmol)2,7-二溴-9-癸基-9-(2-(4-氨基-1,8-萘酰亚胺-9-)乙基-1-)芴溶于10毫升二甲亚砜,再向溶液中加入0.040g(1mmol)60%的氢化钠,电磁搅拌下于0℃反应10分钟,然后向体系中加入0.71g(5.0mmol)碘甲烷,反应3小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9-癸基-9-(2-(4-二甲基氨基-1,8-萘酰亚胺-9-)乙基-1-)芴0.356g,产率92%。
实施例3:2,7-二溴-9,9-(10-(4-氨基-1,8-萘酰亚胺-9-)癸基-1-)芴
在氮气气氛保护下,将2.12g(10.0mmol)4-胺基-1,8-萘酰亚胺溶于60毫升二甲基亚砜,再向溶液中加入0.56g(10.0mmol)粉末化的氢氧化钾,电磁搅拌下于150℃反应十分钟,再向体系中逐渐加入0.381g(5.0mmol)2,7-二溴-9,9-(10-溴癸基-1-)芴,反应14小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9,9-(10-(4-氨基-1,8-萘酰亚胺-9-)癸基-1-)芴2.867g,产率56%。
实施例4:2,7-二溴-9,9-(10-(4-二癸基氨基-1,8-萘酰亚胺-9-)癸基-1-)芴
在氮气气氛保护下,将0.512g(0.5mmol)2,7-二溴-9-癸基-9-(2-(4-氨基-1,8-萘酰亚胺-9-)乙基-1-)芴溶于10毫升二甲亚砜,再向溶液中加入0.40g(10mmol)60%的氢化钠,电磁搅拌下于60℃反应10分钟,然后向体系中加入1.105g(5.0mmol)1-溴癸烷,反应3小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9,9-(10-(4-二癸基氨基-1,8-萘酰亚胺-9-)癸基-1-)芴0.209g,产率32%。
实施例5:2,7-二溴-9-癸基-9-(2-(4-(咔唑基-N-)1,8-萘酰亚胺-9-)乙基-1-)芴的合成
在氮气气氛保护下,将0.362g(1mmol)4-(咔唑基-N-)-1,8-萘酰亚胺溶于100毫升二甲亚砜,再向溶液中加入0.168g(3.0mmol)粉末化的氢氧化钾,电磁搅拌下于90℃反应十分钟,再向体系中逐渐加入0.571g(1.0mmol)2,7-二溴-9-癸基-9-(2-溴乙基-1-)芴。反应14小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9-癸基-9-(2-(4-(咔唑基-N-)1,8-萘酰亚胺-9-)乙基-1-)芴0.315g,产率37%。
实施例6:2,7-二溴-9,9-二(6-(4-(4-甲氧基苯基)1,8-萘酰亚胺-9-)己基-1-)芴的合成
在氮气气氛保护下,将0.606g(2.0mmol)4-(4-甲氧基苯基)-1,8-萘酰亚胺溶于5毫升二甲亚砜,再向溶液中加入0.168g(3.0mmol)粉末化的氢氧化钾,电磁搅拌下于50℃反应十分钟,再向体系中逐渐加入0.52g(0.8mmol)2,7-二溴-9,9-二(6-(溴己基-1-)芴,反应14小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9,9-二(6-(4-(4-甲氧基苯基)1,8-萘酰亚胺-9-)己基-1-)芴0.149g。产率17%。
实施例7:2,7-二溴-9-己基-9-(6-(4-(9,9-二己基芴基-2)-1,8-萘酰亚胺-9-)己基-1-)芴的合成
在氮气气氛保护下,将1.058g(2.0mmol)4-(9,9-二己基芴基-2)-1,8-萘酰亚胺溶于5毫升二甲亚砜,再向溶液中加入0.56g(10.0mmol)粉末化的氢氧化钾,电磁搅拌下于50℃反应十分钟,再向体系中逐渐加入5.71g(10mmol)2,7-二溴-9-己基-9-(6-溴己基-1-)芴,反应1小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9,9-二(6-(4-(4-甲氧基苯基)1,8-萘酰亚胺-9-)己基-1-)芴1.306g。产率64%
实施例8:4-(萘基-2)-1,8-萘酰亚胺的合成
在氮气气氛保护下,将13.76g(80mmol)萘-2-硼酸,13.80g(50mmol)4-溴-1,8-萘酰亚胺,13.80g(100mmol)碳酸钾,50ml水,200ml甲苯,0.231g(0.02mmol)四(三苯基膦)合钯的混合物在80℃搅拌反应24小时,氯仿萃取后反复洗涤,干燥,过滤,除去溶剂后用乙酸乙酯重结晶,得到纯中间产物4-(萘基-2-)-1,8-萘酰亚胺15.02g,产率93%。
实施例9:2,7-二溴-9-己基-9-(6-(4-(萘基-2)-1,8-萘酰亚胺-9-)己基-1-)芴
在氮气气氛保护下,将0.646g(2.0mmol)4-(萘基-2)-1,8-萘酰亚胺溶于5毫升二甲亚砜,再向溶液中加入0.168g(3.0mmol)粉末化的氢氧化钾,电磁搅拌下于20℃反应十分钟,再向体系中逐渐加入5.71g(10mmol)2,7-二溴-9-己基-9-(6-溴己基-1-)芴,反应50小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9-己基-9-(6-(4-(萘基-2)-1,8-萘酰亚胺-9-)己基-1-)芴1.09g。产率67%
实施例10:2,7-二溴-9-丙基-9-(2-(4-甲氧基-1,8-萘酰亚胺-9-)乙基-1-)芴的合成
在氮气气氛保护下,将2.27g(10.0mmol)4-甲氧基-1,8-萘酰亚胺溶于60毫升二甲基亚砜,再向溶液中加入0.84g(15.0mmol)粉末化的氢氧化钾,电磁搅拌下于50℃反应十分钟,再向体系中逐渐加入2.838g(6.0mmol)2,7-二溴-9-丙基-9-(2-溴乙基-1-)芴。反应14小时,氯仿萃取后反复洗涤,干燥,过滤,柱色谱分离产物,得到纯中间产物2,7-二溴-9-己基-9-(2-(4-甲氧基-1,8-萘酰亚胺-9-)乙基-1-)芴2.637g,产率71%。
实施例11:
在氮气保护下,向反应瓶中加入0.2736g(0.499mmmol)9,9-二辛基-2,7-二溴代芴,0.2792g(0.5mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.0007g(0.001mmol)2,7-二溴-9-癸基-9-(2-(4-二甲基氨基-1,8-萘酰亚胺-9-)乙基-1-)芴,0.4140g(3.0mmol)碳酸钾,1.5mL水,7mL甲苯,0.009g Aliquat 336的混合物,在80℃反应十分钟,然后向反应瓶中加入0.0115g(0.01mmol)四(三苯基膦)合钯,100℃搅拌反应12小时,反应混合物用氯仿溶解,三氯化铁水溶液洗一次,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.253g,产率65%。产物性能如下:数均分子量为32,200,将该聚合物在石英基底上旋涂所成的膜吸收光谱和荧光光谱见附图1;.
单层器件(器件结构为:ITO/PEDOT/Polymer/Ca/Al)的组装条件为:采用预先清洗的ITO玻璃为阳极,随后旋涂一层导电高分子-聚噻吩衍生物(PEDOT)(50nm)。PEDOT修饰后的ITO在150℃下真空干燥3小时后,将浓度为10毫克/毫升聚合物的氯仿溶液在转速1500转/分钟的条件下旋涂在ITO表面。随后,在高真空的条件下,蒸镀10nm的金属钙和100nm的金属铝。单层电致发光器件性能如下:启动电压3.8伏,最大亮度6700cd/m2,最大电致发光效率为4.4cd/A,其电致发光光谱见附图2,它的色坐标(0.14,0.20)。
实施例12:
在氮气保护下,向反应瓶中加入0.2194g(0.40mmmol)9,9-二辛基-2,7-二溴代芴,0.2792g(0.50mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.0730g(0.10mmol)2,7-二溴-9-癸基-9-(2-(4-二甲基氨基-1,8-萘酰亚胺-9-)乙基-1-)芴,0.276g(2.0mmol)碳酸钾,1.0mL水,7mL甲苯,0.004g Aliquat 336的混合物,在60℃反应十分钟,然后向反应瓶中加入0.0012g(0.001mmol)四(三苯基膦)合钯,60℃搅拌反应120小时,反应混合物用氯仿溶解,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.215g,产率53%。产物性能如下:数均分子量为26,700,薄膜紫外最大吸收为392nm,固态荧光发射峰值为486nm。
单层器件组装条件同实施例11.单层电致发光器件性能如下:启动电压11.6伏,最大亮度711cd/m2,最大电致发光效率为0.74cd/A,色坐标为(0.20,0.48)。
实施例13:
在氮气保护下,向反应瓶中加入0.2792g(0.500mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.3514g(0.500mmol)2,7-二溴-9-癸基-9-(2-(4-二甲基氨基-1,8-萘酰亚胺-9-)乙基-1-)芴,0.4140g(3.0mmol)碳酸钾,1.5mL水,6mL甲苯的混合物,在80℃反应十分钟,然后向反应瓶中加入0.0115g(0.01mmol)四(三苯基膦)合钯,50℃搅拌反应120小时,反应混合物用氯仿溶解,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.226g,产率47%。产物性能如下:数均分子量为15,400,薄膜紫外最大吸收为397nm,固态荧光发射峰值为517nm。
单层器件组装条件同实施例11.单层电致发光器件性能如下:启动电压8.6伏,最大亮度543cd/m2,最大电致发光效率为0.78cd/A,色坐标为(0.26,0.58)。
实施例14:
在氮气保护下,向反应瓶中加入0.2739g(0.4995mmmol)9,9-二辛基-2,7-二溴代芴,0.2792g(0.5mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.0007g(0.0005mmol)2,7-二溴-9,9-(10-(4-二癸基氨基-1,8-萘酰亚胺-9-)癸基-1-)芴,1.38g(10.0mmol)碳酸钾,5mL水,7mL甲苯,0.21g Aliquat 336的混合物,在80℃反应十分钟,然后向反应瓶中加入0.0115g(0.01mmol)四(三苯基膦)合钯,搅拌回流反应12小时,反应混合物用氯仿溶解,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.252g,产率65%。产物性能如下:数均分子量为31,000,薄膜紫外最大吸收为392nm,固态荧光发射峰值为468nm;
单层器件组装条件同实施例11.单层电致发光器件性能如下:启动电压3.5伏,最大亮度6641cd/m2,最大电致发光效率为2.58cd/A,色坐标为(0.16,0.18);
实施例15:
在氮气保护下,向反应瓶中加入0.2632g(0.48mmmol)9,9-二辛基-2,7-二溴代芴,0.2792g(0.5mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.0174g(0.02mmol)2,7-二溴-9-癸基-9-(2-(4-(咔唑基-N-)1,8-萘酰亚胺-9-)乙基-1-)芴,0.4140g(3.0mmol)碳酸钾,1.5mL水,7mL甲苯,0.08g Aliquat336的混合物,在80℃反应十分钟,然后向反应瓶中加入0.0115g(0.01mmol)四(三苯基膦)合钯,100℃搅拌反应12小时,反应混合物用氯仿溶解,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.253g,产率65%。产物性能如下:数均分子量为24,000,薄膜紫外最大吸收为392nm,固态荧光发射峰值为481nm;
单层器件组装条件同实施例11,单层电致发光器件性能如下:启动电压7.6伏,最大亮度341cd/m2,最大电致发光效率为0.38cd/A,色坐标为(0.20,0.26);
实施例16:
在氮气保护下,向反应瓶中加入0.2740g(0.4998mmmol)9,9-二辛基-2,7-二溴代芴,0.2792g(0.5mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.0002g(0.0002mmol)2,7-二溴-9,9-二(6-(4-(4-甲氧基苯基)1,8-萘酰亚胺-9-)己基-1-)芴,0.4140g(3.0mmol)碳酸钾,1.5mL水,7mL甲苯,0.08g Aliquat336的混合物,在80℃反应十分钟,然后向反应瓶中加入0.0115g(0.01mmol)四(三苯基膦)合钯,90℃搅拌反应12小时,反应混合物用氯仿溶解,三氯化铁水溶液洗一次,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.221g,产率57%。产物性能如下:数均分子量为32,800,薄膜紫外最大吸收为392nm,固态荧光发射峰值为421nm。
单层器件组装条件同实施例11.单层电致发光器件性能如下:启动电压11.6伏,最大亮度541cd/m2,最大电致发光效率为1.21cd/A,色坐标为(0.16,0.12)
实施例17:
在氮气保护下,向反应瓶中加入0.2725g(0.497mmmol)9,9-二辛基-2,7-二溴代芴,0.2792g(0.5mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.0031g(0.003mmol)2,7-二溴-9-己基-9-(6-(4-(9,9-二己基芴基-2)-1,8-萘酰亚胺-9-)己基-1-)芴,0.4140g(3.0mmol)碳酸钾,1.5mL水,7mL甲苯,0.08gAliquat 336的混合物,在80℃反应十分钟,然后向反应瓶中加入0.0115g(0.01mmol)四(三苯基膦)合钯,100℃搅拌反应12小时,反应混合物用氯仿溶解,三氯化铁水溶液洗一次,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.253g,产率65%。产物性能如下:数均分子量为32,000,薄膜紫外最大吸收为392nm,固态荧光发射峰值为443nm。
单层器件组装条件同实施例11.单层电致发光器件性能如下:启动电压4.6伏,最大亮度4623cd/m2,最大电致发光效率为3.64cd/A,色坐标为(0.16,0.15)
实施例18:
在氮气保护下,向反应瓶中加入0.2736g(0.499mmmol)9,9-二辛基-2,7-二溴代芴,0.2792g(0.5mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.0008g(0.001mmol)2,7-二溴-9-己基-9-(6-(4-(萘基-2)-1,8-萘酰亚胺-9-)己基-1-)芴,0.4140g(3.0mmol)碳酸钾,1.5mL水,7mL甲苯,0.08g Aliquat 336的混合物,在80℃反应十分钟,然后向反应瓶中加入0.115g(0.1mmol)四(三苯基膦)合钯,80℃搅拌反应12小时,反应混合物用氯仿溶解,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.279g,产率72%。产物性能如下:数均分子量为37,800,薄膜紫外最大吸收为392nm,固态荧光发射峰值为435nm。
单层器件组装条件同实施例11.单层电致发光器件性能如下:启动电压3.6伏,最大亮度4520cd/m2,最大电致发光效率为1.78cd/A,色坐标为(0.16,0.14)
实施例19:
在氮气保护下,向反应瓶中加入0.2736g(0.499mmmol)9,9-二辛基-2,7-二溴代芴,0.2792g(0.5mmol)9,9-二辛基-2,7-(三亚甲基硼酸酯基)芴,0.0006g(0.001mmol)2,7-二溴-9-丙基-9-(2-(4-甲氧基-1,8-萘酰亚胺-9-)乙基-1-)芴,0.4140g(3.0mmol)碳酸钾,1.5mL水,7mL甲苯,0.08g Aliquat 336的混合物,在80℃反应十分钟,然后向反应瓶中加入0.0115g(0.01mmol)四(三苯基膦)合钯,120℃搅拌反应12小时,反应混合物用氯仿溶解,三氯化铁水溶液洗一次,水洗多次,干燥,浓缩,然后用甲醇沉降三次,将产物置于索氏提取器,用丙酮抽提24小时,然后用氯仿溶解后在甲醇中沉降。产物真空干燥得淡黄色固体0.253g,产率65%。产物性能如下:数均分子量为22,600,薄膜紫外最大吸收为392nm,固态荧光发射峰值为440nm。
单层器件组装条件同实施例11.单层电致发光器件性能如下:启动电压4.6伏,最大亮度2141cd/m2,最大电致发光效率为1.28cd/A,色坐标为(0.16,0.13)。
蓝色电致发光高分子聚芴类材料及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0