

IPC分类号 : C07B53/00,C07D239/54,C07D239/553,C07D405/06






专利摘要
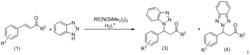
本发明公开了一种通过共轭加成‑质子化反应合成含硫侧链的手性嘧啶非环核苷的方法。以奎宁‑硫脲催化剂,将硫代乙酸和和双键底物3在醚溶剂或甲苯中反应,得到含硫侧链的手性嘧啶非环核苷4;该反应通过使用特定的手性催化剂和反应条件,能以高产率和高对应选择性得到含硫的手性产物。该反应具有原料易得、反应条件温和、催化剂绿色环保等优点,为合成手性嘧啶非环核苷类似物提供了一条简洁实用的合成方法。
权利要求
1.一种通过不对称共轭加成-质子化的串联反应合成手性嘧啶非环核苷的方法,其特征在于,包括如下步骤:以奎宁-硫脲催化剂,将硫代乙酸和双键底物3在醚溶剂中反应,得到含硫侧链的手性嘧啶非环核苷4;反应方程式如下:
R选自:甲基、乙基、氢、卤素或三氟甲基;Pg选自:苯甲酰基、4-甲氧基苯甲酰基、3,4-二甲氧基苯甲酰基、胡椒酰基或萘甲酰基;所述奎宁-硫脲催化剂选自
2.根据权利要求1中一种通过不对称共轭加成-质子化的串联反应合成手性嘧啶非环核苷的方法,其特征在于:所述底物3中R基团卤素选自F,Cl,Br或I。
3.根据权利要求1中一种通过不对称共轭加成-质子化的串联反应合成手性嘧啶非环核苷的方法,其特征在于:所述奎宁-硫脲催化剂的加入量为底物3的1-10mol%。
4.根据权利要求1中一种通过不对称共轭加成-质子化的串联反应合成手性嘧啶非环核苷的方法,其特征在于:所述反应温度为-40℃至0℃。
5.根据权利要求1中一种通过不对称共轭加成-质子化的串联反应合成手性嘧啶非环核苷的方法,其特征在于:所述硫代乙酸与双键底物3摩尔比为1-1.5:1。
说明书
技术领域
本发明属于有机合成化学技术领域,具体涉及一种通过共轭加成-质子化反应合成含硫侧链的手性嘧啶非环核苷的方法。
背景技术
非环核苷类药物在抗病毒与抗肿瘤药物中占据着非常重要的地位,在近几十年的药物研发中,非环核苷领域贡献出了多种有效药物。如今,大量的资源也都不断投入到非环核苷类新药的研发中,在目前上市的和处在临床阶段的药物中,非环核苷类药物也占据着想到高的比例。但是目前市面上的非环核苷类药物还都普遍存在着不良反应较多、生物利用度低、容易产生耐药性和代谢过快等等问题。因此研究新型的非环核苷类药物势在必行。
目前传统的手性嘧啶非环核苷的合成方法主要是通过嘧啶碱基和手型原料反应从而生成手性的嘧啶非环核苷类化合物。但是,这种方法存在着手性原料合成困难,步骤多,对环境有较大影响等问题。并且,随着社会对于新型非环核苷类药物的需求逐渐增大,更加科学、高效的合成新型非环核苷类化合物成的研究刻不容缓。
与此同时,许多含硫化合物也显示出了很好的生物活性,到目前为止,侧链上含硫取代的手性嘧啶非环核苷尚未见报道。因此寻求一种简便、绿色、高效的不对称共轭加成-质子化反应来合成含硫侧链的手性嘧啶非环核苷类似物,立足于解决此类化合物合成过程中原料昂贵,过程复杂的问题,为核苷类药物的合成及应用提供参考价值,满足新型抗病毒及抗肿瘤药物的研究对原料的需求就显得非常重要。
发明内容
为了解决现有技术的不足,本发明提供了一种通过共轭加成-质子化反应合成含硫侧链的手性嘧啶非环核苷的方法。
一种通过不对称共轭加成-质子化的串联反应合成手性嘧啶非环核苷的方法,技术方案如下:以奎宁-硫脲催化剂,将硫代乙酸和和双键底物3在醚溶剂或甲苯中反应,得到含硫侧链的手性嘧啶非环核苷4;反应方程式如下:
R选自:甲基、乙基、氢、卤素或三氟甲基;Pg选自:苯甲酰基、4-Cl苯甲酰基、4-甲氧基苯甲酰基、3,4-二甲氧基苯甲酰基、胡椒酰基或萘甲酰基。
进一步地,在上述技术方案中,底物3中R基团中的卤素选自F,Cl,Br或I。
进一步地,在上述技术方案中,奎宁-硫脲催化剂选自C3和C5:
具体结构式为:
进一步地,在上述技术方案中,所述奎宁-硫脲催化剂的加入量为底物3的1-10mol%。
进一步地,在上述技术方案中,所述反应温度为低温,优选-40℃至0℃。
进一步地,在上述技术方案中,所述醚溶剂选自乙醚、环戊基甲醚或乙二醇二甲醚。
进一步地,在上述技术方案中,所述硫代乙酸与双键底物3摩尔比为1-1.5:1。
进一步地,在上述技术方案中,所述反应中添加4A分子筛。
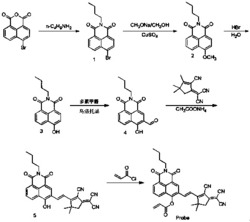
进一步地,在上述技术方案中,所述双键底物3采用如下方法合成:氮气保护下,将嘧啶(10.0mmol),三苯基膦(26mg),醋酸钠(16mg),室温加入到50mL甲苯中,再加入丙炔酸乙酯(1.2mL),升温回流反应后得到双键底物3:
其中:R选自下列基团中的一种:甲基、乙基、氢、卤素(F,Cl,Br,I)、三氟甲基、对甲苯基、4-苯乙炔基;Pg选自下列基团中的一种:苯甲酰基、4-Cl苯甲酰基、4-甲氧基苯甲酰基、3,4-二甲氧基苯甲酰基、胡椒酰基、萘甲酰基。
当R为对甲苯基时,还需要再将碘代物与对甲苯硼酸在钯催化下再次偶联后得到,反应方程式如下:
当R为4-苯乙炔基时,还需要再将碘代物与苯乙炔在钯催化下再次偶联后得到,反应方程式如下:
以上合成方法得到的底物3的具体结构如下:
发明的有益效果:
本发明中原料易得,操作简单,反应时间短,催化效率高。该反应通过使用特定的手性催化剂和反应条件,能以高产率和高对应选择性得到含硫的手性产物。该反应具有原料易得、反应条件温和、催化剂绿色环保等优点,为合成手性嘧啶非环核苷类似物提供了一条简洁实用的合成方法。
具体实施方式
实施例1
底物合成和反应条件筛选
在圆底烧瓶中加入3-位保护的嘧啶底物1(10mmol,1.0equiv)、PPh3(0.2mmol,0.02equiv),再加入醋酸钠(2mmol,0.2equiv)和30ml经过无水甲苯,然后用移液枪加入冰醋酸(5mmol,0.5equiv)搅拌均匀后加入丙炔酸酯(12mmol,1.2equiv),然后将反应置于105℃油浴锅中并置换氮气,反应搅拌过夜。反应结束后将反应移出油浴锅并冷却至室温,然后向反应液中加入80mL乙酸乙酯,用水萃取三次,经过干燥杯干燥过滤后用旋转蒸发仪温度60℃旋干剩余溶剂,并加入硅胶搅拌后用石油醚和乙酸乙酯体系过柱,即可得到嘧啶1位丙烯酸酯取代的底物3。
共轭加成-质子化反应合成含硫侧链的手性嘧啶非环核苷反应条件筛选:以底物3a和硫代乙酸反应得到4a为例:
反应方程式如下:
[a]Unless otherwise noted,the reaction conditions were:in a testtube,3a(0.05 mmol),thiolacetic acid(0.06 mmol),and catalyst were adde d,followed by the addition of solvent(0.4 mL)at-20 ℃ for 15 min.[b]Isolatedyield.[c]Determined by chiral HPLC analysis.[d]The catal yst C3(10mol%)and 2(0.06mmol)was dissolved in solvent(0.2mL),then 3a(0.05mmol)in solvent(0.2mL)was added dropwise in 20min.[e]Reaction time:24h.[f] MS(25mg)was added.DMB=dimethylbenzene;MTBE=methyl tert-butyl ether;EDGE=ethylene gly col dimethylether.
实施例2
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL乙醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。该产物通过X-ray单晶衍射确定了其手性的绝对构型为S。收率99%,ee:99%。[α]
实施例3
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率99%,ee:80%。[α]
实施例4
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4ml环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率99%,ee:83%。[α]
实施例5
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5ml二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率99%,ee:83%。
实施例6
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率99%,ee:74%。
实施例7
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率99%,ee:88%。
实施例8
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率99%,ee:82%。[α]
实施例9
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率95%,ee:82%。[α]
实施例10
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率95%,ee:77%。[α]
实施例11
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率99%,ee:61%。[α]
实施例12
反应管中加入25mg 4A分子筛,然后将奎宁硫脲催化剂(0.9mg,0.0015mmol)溶解于0.4mL环戊基甲醚加入到反应管内,接下来用移液枪将(3.6μL,0.05mmol)的硫代乙酸加入反应管内然后将反应管置于-20℃冰浴中搅拌15分钟。接下来将双键底物(0.05mmol)以固体形式加入反应中溶剂中继续搅拌15分钟。反应完成后在反应管中加入5mL二氯甲烷,并用水萃取三次,然后经过干燥杯过滤干燥后加入硅胶旋干剩余溶剂,并用乙酸乙酯和石油醚体系过柱子得到反应产物。收率98%,ee:85%。[α]
实施例13
采用与实施例12同样的反应条件,仅将反应底物进行更换,得到的产物收率和对映选择性如下:
以上实施例描述了本发明的基本原理、主要特征及优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
一种通过共轭加成-质子化反应合成含硫侧链的手性嘧啶非环核苷的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。





![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)






动态评分
0.0