










专利摘要
一种具有抗肿瘤活性的齐墩果酸化学修饰物及其制备方法,涉及一种天然产物齐墩果酸的结构改造方法,该方法通过天然产物齐墩果酸的化学结构改造及修饰,得到一系列具有生物活性的结构类似物。经过药理实验证明,该齐墩果酸化学修饰物对人体宫颈癌Hela细胞,人肝癌HepG2细胞和胃癌BGC-823细胞具有较好的抑制作用,且优于母体化合物齐墩果酸。所述齐墩果酸化学修饰物包括以下三类。
权利要求
1.一种具有抗肿瘤活性的齐墩果酸化学修饰物,其特征在于,所述修饰物为对A环,C环的结构改造,以及对C-3位和C-28位的修饰,得到齐墩果酸酯类或酰胺类化合物;所述齐墩果酸化学修饰物包括以下三类:
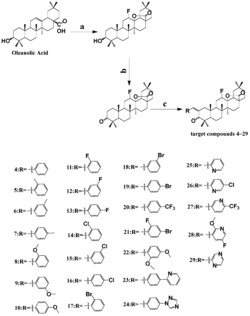
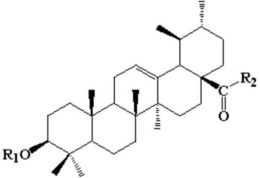
(1)在齐墩果酸结构的基础上进行修饰,C-3位为氢,烷氧基,C-12,13位引入双羟基或烷氧基,C-28位用烷氧基,羟基,胺基取代,得到一系列化合物;化合物结构如下表所示:
(2)在齐墩果酸结构的基础上,将A环C-3位修饰为3-肟基或3-肉桂酰氧亚氨基,同时在C-28位引入酯基或胺基,得到一系列化合物,结构如下表所示:
(3)在齐墩果酸结构的基础上,将A环扩环为七元内酰胺环,C-28位羧基引入不同的烷氧基;
一种抗肿瘤活性的齐墩果酸化学修饰物制备方法,其特征在于,所述方法包括以下步骤:
(1)齐墩果酸C-12位双键被间氯过氧苯甲酸氧化,生成环氧化合物,在酸性条件下开环得到二羟基化合物;最终制得3β,12α,13β-三羟基-齐墩果烷型-28-羧酸(OA-1);
(2)OA-1再与相应的卤代烷和碳酸钾溶解于DMF中,最后反应生成3β,12α,13β-三羟基-齐墩果烷型-28-羧酸酯类化合物I1~I2;
(3)OA-1与乙酸酐在吡啶催化下,3,12位羟基被乙酰化,最终生成3β,12α-二乙酰氧基-13β-羟基-齐墩果烷型-28-羧酸(OA-2);
(4)OA-2与草酰氯反应后,再与相应胺反应生成酰胺类目标化合物I3~I4;
(5)齐墩果酸和琼斯试剂反应得到3-氧代齐墩果酸(OA-3);
(6)OA-3溶于适量吡啶中,与盐酸羟按反应得到3-肟基-齐墩果烷型-12-烯-28-羧酸(OA-4);
(7)OA-4与相应的卤代烷按照步骤(2),得到目标化合物II1~II2;
(8)肉桂酸与草酰氯反应,制得肉桂酰氯;
(9)OA-4与肉桂酰氯溶解于苯中,加入少量DMAP,反应得到3-肉桂酰氧亚氨基-齐墩果烷型-12-烯-28-羧酸(OA-5);再按照步骤(2)与1,2-二溴乙烷反应得到3-肉桂酰氧亚氨基-齐墩果烷型-12-烯-28-(2-溴-1-)乙脂II3;
(10)OA-5与相应的胺按照步骤(4)反应得到目标化合物II4~II7;
(11)OA-4在二氧六环中与二氯亚砜反应,经贝克曼重排成七元内酰胺衍生物A-同环-4-氮杂-3-氧代齐墩果烷型-12-烯-28-羧酸III1;
(12)III1与相应的卤代烷按照步骤(2),得到目标化合物III2~III3。
说明书
技术领域
本发明涉及一种天然产物齐墩果酸的结构改造方法,特别是涉及一种具有抗肿瘤活性的齐墩果酸化学修饰物及其制备方法。
背景技术
齐墩果酸(OleanolicAcid,OA),又名庆四素,为五环三萜类化合物,以游离或结合成苷的形式广泛存在于白花蛇舌草、山楂、丁香、大枣、女贞子、枇杷叶、木及夏枯草等植物中。化学名为(3β)-3-Hydroxyolean-12-en-28-oicacid,纯品为白色结晶性粉末,无臭、无味。分子式为:C30H48O3,分子量:456.71。熔点:308℃~310℃,[α]20D+68°~+78°(C=0.15,氯仿)。不溶于水,可溶于乙醇、氯仿、丙酮。近年来研究发现,齐墩果酸具有广泛的生物活性,包括降糖降脂作用,抗肿瘤,抗病毒及微生物,抗衰老,抗溃疡,消炎,对免疫系统的影响,保肝护肝等。
齐墩果酸的化学结构式:
近年来,齐墩果酸对肿瘤的抑制和杀伤效应日益受到重视。齐墩果酸能够抑制肿瘤的形成,阻碍肿瘤诱发和诱导肿瘤细胞分化,且能有效地抑制血管发生肿瘤细胞的侵害和转移。对肿瘤细胞表现出较强的细胞毒性,如人肺纤维原细胞WI-38、人肝癌细胞株HepG2、结肠癌HCTl5细胞株、卵巢癌HO-8910细胞株、乳腺癌MCF-7细胞株等。对恶性血液病细胞株K562、HI,60、Jurkat等的增殖活性也同样具有抑制作用。
发明内容
本发明的目的在于提供一种具有抗肿瘤活性的齐墩果酸化学修饰物及其制备方法,该方法以齐墩果酸为先导化合物,设计出一系列齐墩果酸化学修饰物,该类化合物对人体宫颈癌Hela细胞,人肝癌HepG2细胞和胃癌BGC-823细胞具有较好的抑制活性。
本发明的目的是通过以下技术方案实现的:
一种具有抗肿瘤活性的齐墩果酸化学修饰物,主要对齐墩果酸A环,C环的结构改造,以及对C-3位和C-28位的化学修饰,得到齐墩果酸酯类或酰胺类化合物。所述齐墩果酸化学修饰物包括以下三类:
(1)在齐墩果酸结构的基础上进行修饰,在C-3位引入氢,烷氧基,C-12,13位引入双羟基或烷氧基,C-28位用烷氧基,羟基,胺基取代,得到一系列化合物。化合物结构如下表所示:
(2)在齐墩果酸结构的基础上进行修饰,将A环C-3位修饰为3-肟基或3-肉桂酰氧亚氨基,同时在C-28位引入酯基或酰胺基,得到一系列化合物,化合物结构如下表所示:
(3)在齐墩果酸结构的基础上进行修饰,将A环扩环为七元内酰胺环,C-28位羧基引入不同的烷氧基。得到一系列化合物,化合物结构如下表所示:
一种具有抗肿瘤活性的齐墩果酸化学修饰物制备方法,所述方法包括以下步骤:
(1)齐墩果酸C-12位双键被间氯过氧苯甲酸氧化,生成环氧化合物,在酸性条件下开环得到二羟基化合物。最终制得3β,12α,13β-三羟基-齐墩果烷型-28-羧酸(OA-1)。
(2)OA-1再与相应的卤代烷和碳酸钾溶解于DMF中,反应生成3β,12α,13β-三羟基-齐墩果烷型-28-羧酸酯类化合物I1~I2。
(3)OA-1与乙酸酐在吡啶催化下,3,12位羟基被乙酰化,生成3β,12α-二乙酰氧基-13β-羟基-齐墩果烷型-28-羧酸(OA-2)。
(4)OA-2与草酰氯反应后,与相应胺反应生成3β,12α-二乙酰氧基-13β-羟基-齐墩果烷型-28-酰胺类目标化合物I3~I4。
(5)齐墩果酸和琼斯试剂反应得3-氧代齐墩果酸(OA-3)。
(6)OA-3溶于适量吡啶中,与盐酸羟按反应得到3-肟基-齐墩果烷型-12-烯-28-羧酸(OA-4)。
(7)OA-4与相应的卤代烷按照步骤(2),得到3-肟基-齐墩果烷型-12-烯-28-羧酸酯类目标化合物II1~II2。
(8)肉桂酸与草酰氯反应得肉桂酰氯。
(9)OA-4与肉桂酰氯溶解于苯中,加入少量DMAP,反应得到3-肉桂酰氧亚氨基-齐墩果烷型-12-烯-28-羧酸(OA-5)。再按照(2)步骤与1,2-二溴乙烷反应得到3-肉桂酰氧亚氨基-齐墩果烷型-12-烯-28-(2-溴-1-)乙脂II3。
(10)OA-5与相应的胺按照(4)步骤反应得到3-肉桂酰氧亚氨基-齐墩果烷型-12-烯-28-酰胺类目标化合物II4~II7。
(11)OA-4在二氧六环中与二氯亚砜反应,经贝克曼重排成七元内酰胺衍生物A-同环-4-氮杂-3-氧代齐墩果烷型-12-烯-28-羧酸III1。
(12)III1与相应的卤代烷按照步骤(2),得到A-同环-4-氮杂-3-氧代齐墩果烷型-12-烯-28-羧酸酯类目标化合物III2~III3。
本发明的优点与效果是:
本发明对天然产物齐墩果酸进行化学修饰,得到一系列齐墩果酸的结构类似物,经过药理实验证明,它们对人体宫颈癌Hela细胞,人肝癌HepG2细胞和胃癌BGC-823细胞具有较好的抑制作用,且优于母体化合物齐墩果酸。
具体实施方式
下面结合实例,对本发明做进一步详述。
1.女贞子的乙醇提取浸膏以石油醚、1%氢氧化钠和水洗至洗出液无色,无水乙醇加热溶解,活性炭脱色,经凝析分离、纯化精制得白色晶体齐墩果酸(OA)。
2.齐墩果酸的C-12位双键被间氯过氧苯甲酸氧化,生成环氧化合物,在酸性条件下开环,最后得到3β,12α,13β-三羟基-齐墩果烷型-28-羧酸(OA-1),在OA-1合成线路的基础上,C-28羧酸与相应的卤代烷在碳酸钾碱性条件下,溶解于DMF中,得到3β,12α,13β-三羟基-齐墩果烷型-28-羧酸酯类化合物I1~I2。
其中:R2为-OCH(CH3)2和-OCH2CH(CH3)2。
OA-1与乙酸酐在吡啶催化下,3,12位羟基被乙酰化,得到3β,12α-二乙酰氧基-13β-羟基-齐墩果烷型-28-羧酸(OA-2),在OA-2合成线路的基础上与草酰氯反应得到酰氯后,再与相应胺反应得到目标化合物I3~I4。
其中:R2为-OC2H5和 。
肉桂酸与草酰氯反应,制得肉桂酰氯。OA-4与肉桂酰氯溶解于苯中,加入少量DMAP,反应得到3-肉桂酰氧亚氨基-齐墩果烷型-12-烯-28-羧酸(OA-5)。再与1,2-二溴乙烷在碳酸钾碱性条件下,溶解于DMF中,反应得到目标化合物II3。
OA-5与草酰氯反应得到酰氯后,再与相应胺反应得到目标化合物II4~II7。
其中:R5为-N(C2H5)2, 。
OA-4在二氧六环中与二氯亚砜反应,经贝克曼重排成七元内酰胺衍生物A-同环-4-氮杂-3-氧代齐墩果烷型-12-烯-28-羧酸III1。III1与相应的卤代烷在碳酸钾碱性条件下,溶解于DMF中,反应得到目标化合物III2~III3。
其中:R6为-OH,-OC2H5和 。
以吉非替尼和VP-16为阳性对照物,采取MTT法对齐墩果酸及其所合成的化合物进行初步的体外抗肿瘤活性测试。研究表明部分化合物对人体宫颈癌Hela细胞,人肝癌HepG2细胞和胃癌BGC-823细胞具有较好的抑制活性,且强于齐墩果酸。化合物结构以及体外测试结果如下表所示。
注;a.化合物浓度在10-5mol/L时测得的抑制率,b.IC50表示半数有效抑制浓度。
下面结合实施例对本发明做进一步说明:
实施例1
3β,12α,13β-三羟基-齐墩果烷型-28-羧酸异丙酯(I1)的制备:
将起始原料OA(l.9mmol,0.87g)与间氯过氧苯甲酸(3.8mmol,0.65g)溶解在10mL二氯甲烷中。反应混和液室温搅拌过夜。加入10mL水,分层后,水层以二氯甲烷萃取(10mL×2)。合并有机相,以饱和碳酸氢钠溶液洗至微碱性,再水洗至中性。无水硫酸镁干燥,过滤、浓缩,得到环氧化物。然后将环氧化物(0.4mmol)溶于10mL氯仿中,加入适量浓盐酸调PH至中性。反应混和液室温搅拌6小时。加入10mL水,分层后,水层以氯仿萃取(10mL×3)。合并有机相,饱和碳酸氢钠溶液洗涤至微碱性,再水洗至中性。无水硫酸镁干燥,过滤,浓缩,真空干燥得到白色无定形固体OA-1。
在25mL的茄形瓶中加入化合物OA-1(0.100g,0.20mmol),无水碳酸钾0.06g,N,N-二甲基甲酰胺4mL,缓慢滴加溴代异丁烷3滴(0.88mmol),室温反应,TLC检测反应终点。反应结束后,减压蒸出剩余的卤代烷,加入适量饱和NaCl溶液2mL,乙酸乙酯萃取(4mL×3),合并有机相,无水硫酸镁干燥,过滤,浓缩,真空干燥得白色固体。经硅胶柱色谱纯化,洗脱剂为石油醚/乙酸乙酯=7/1(V/V),得白色晶状固体I155.8mg,产率50.1%。mp207.2~212.4℃。IR(KBr):3559,2955,1712,1577,1386cm-1。1H-NMR(CDCl3,300MHz)δ:5.21(brs,1H,12-OH),4.93(brs,1H,13-OH),4.65(brs,1H,3-OH),4.20(m,1H,OCH(CH3)2),3.89~3.96(m,1H,H-12),3.12~3.24(m,1H,H-3),1.32(d,J=8.0Hz,3H,COOCH(CH3a)2),1.27(d,J=8.0Hz,3H,COOCH(CH3b)2),1.20,1.15,1.07,0.98,0.90,0.84,0.77(7s,21H,CH3×7)。ESI-MS:533.4(M+H)+。
实施例2
3β,12α-二乙酰氧基-13β-羟基-齐墩果烷型-28-环己胺(I3)的制备:
在50mL单口瓶中加入OA-1(200mg,0.41mmol),四氢呋喃20mL,OA-1溶解后于室温下加入吡啶1mL、乙酸酐(1.35g,13.2mmol)和少量DMAP,室温下搅拌,以TLC检测反应终点(展开剂:石油醚/乙酸乙酯=3/1,显色剂为10%硫酸乙醇溶液),反应完毕,减压蒸除反应溶剂,以水分散固体,2mol/L盐酸调pH至3~4,析出白色固体,抽滤,滤饼水洗至中性,室温自然干燥,得3β,12α-二乙酰氧基-13β-羟基-齐墩果烷型-28-羧酸,粗品经硅胶柱色谱分离纯化,得白色固体(OA-2)189.7mg,产率84.5%。
将化合物OA-2(100mg,0.18mmol)溶解在3mL二氯甲烷中,加入草酰氯(0.8mmol),室温搅拌20小时,生成3β,12α-二乙酰氧基-13β-羟基-齐墩果烷型-28-酰氯,蒸除反应溶剂和未反应的草酰氯,残余物加入2mL环己烷,减压蒸除环己烷,反复操作2次。酰氯中加入2mL二氯甲烷,加三乙胺调pH为9~10,搅拌5分钟后,加入环己胺(0.6mmol),室温下反应,以TLC检测反应终点。反应结束后,减压蒸除二氯甲烷,向反应液中加入3mL饱和NaCl溶液,以2mol/L盐酸调pH至3~4,析出白色固体,抽滤,水洗滤饼至中性,室温干燥,粗品经硅胶柱色谱分离纯化,洗脱剂为石油醚/乙酸乙酯=8/1(V/V),得白色晶体I342.5mg,产率35.6%。mp219.7~225.1℃。IR(KBr):3382,2932,1734,1662,1243cm-1。1H-NMR(CDCl3,300MHz)δ:5.62(brs,1H,NH),4.80(brs,1H,13-OH),3.71(m,1H,NHCH),3.32~3.50(m,1H,H-12),3.19~3.24(m,1H,H-3),2.09(s,3H,CH3CO),2.03(s,3H,CH3CO),1.99~1.94(m,4H,CHCH2×2),1.80~1.75(m,4H,CHCH2CH2×2),1.66~1.60(m,2H,CH2),1.25,1.16,1.07,0.98,0.89,0.93,0.77(21H,7s,CH3×7)。ESI-MS:691.1(M+Cl)+。
实施例3
3-肟基-齐墩果烷型-12-烯-28-乙酯(II1)的制备:
将化合物OA-3(50mg,0.11mmol)溶解于5ml无水吡啶中,加入盐酸羟胺100mg,在120℃回流反应1小时,冷却,用水稀释,减压抽滤,滤饼用水洗,干燥,得到白色粉末3-肟基-齐墩果烷型-12-烯-28-羧酸(OA-4)45.8mg,收率为81.5%。IR(KBr):3244,2944,1685,1648,1460,1386,1365,1278,1179cm-1。
将化合物OA-4(100mg,0.20mmol),溴乙烷(0.06ml)和碳酸钾(121mg,0.8774mmol)溶解于6ml的DMF溶液中,在室温下搅拌过夜。TLC跟踪检测。反应结束后,减压蒸出剩余的卤代烷,加入适量饱和NaCl溶液2mL,乙酸乙酯萃取(4mL×3),合并有机相,无水硫酸镁干燥,过滤,浓缩,真空干燥得白色固体。粗品经硅胶柱色谱分离纯化,洗脱剂为石油醚/乙酸乙酯=4/1(V/V),得白色晶体II166.8mg,产率65.2%。mp189.6~193.5℃;IR(KBr):3263,2943,1723,1633,1462,1385,1363,1263,1181cm-1;1H-NMR(CDCl3,300MHz)δ:5.20(t,J=3.5Hz,1H,H-12),4.08~4.05(m,2H,COOCH2CH3),2.56(d,J=7.5Hz,1H,H-18),1.53(t,J=7.5Hz,1H,H-9),1.25(t,J=8.7Hz,3H,COOCH2CH3),1.10,0.98,0.88,0.86,0.83,0.75,0.68(7s,21H,CH3×7)。ESI-MSm/z:497.5M+。
实施例4
N-[3-肉桂酰氧亚氨基-齐墩果烷型-12-烯-28-酰基]-甲基哌嗪(II5)的制备:
将化合物OA-5(100mg,0.20mmol)溶解在4mL二氯甲烷中,加入草酰氯(0.44mmol),室温搅拌20小时,生成3-肉桂酰氧亚氨基-齐墩果烷型-12-烯-28-酰氯,蒸除反应溶剂和未反应的草酰氯,残余物加入环己烷,减压蒸除环己烷(2mL×3)。后加入2mL二氯甲烷使之完全溶解后,加三乙胺调pH为9~10,搅拌5分钟后,加入N-甲基哌嗪(20mmol),室温下反应6小时,以TLC检测反应终点。反应结束后,减压除溶剂,水分散固体,以2mol/L盐酸调pH至3~4,抽滤,得棕色固体。粗品用硅胶柱色谱纯化,洗脱剂为石油醚/乙酸乙酯=8/1(V/V),得到白色粉末II521mg,产率14%。mp192.8~194.1℃,IR(KBr):1742,1656,1454,1210,721cm-1;1H-NMR(CDCl3,300MHz)δ:7.74(d,2H,Ph-H×2,J=12.0Hz),7.70(d,1H,Ph-CH,J=15.0Hz),7.40(t,2H,Ph-H,J=15.0Hz),6.50(d,1H,CHCOON,J=16.5Hz),5.18(t,J=3.5Hz,1H,H-12),3.40(t,J=7.5Hz,4H,CH2aNHCH2a),3.32(t,J=8.0Hz,4H,CH2bNHCH2b),2.43(d,J=8.0Hz,1H,H-18),2.28(s,3H,NCH3),1.48(t,J=7.5Hz,1H,H-9),1.20,1.16,1.10,0.99,0.94,0.87,0.76(7s,21H,CH3×7).ESI-MS:682.5(M+H)+。
实施例5
A-同环-4-氮杂-3-氧代-齐墩果烷型-12-烯-28-乙酯(III2)的制备:
将化合物OA-4(50mg,0.20mmol)溶于10ml无水二氧六环中,在10℃下滴加新制的二氯亚砜溶液0.2ml,搅拌10min,用10%的氢氧化钾溶液稀释,减压抽滤,滤饼用水洗,干燥,得到白色固体(III1)32.8mg,产率75.6%。
将在50mL的茄形瓶中加入化合物III1(100mg,0.20mmol),无水碳酸钾0.06g,N,N-二甲基甲酰胺4mL,缓慢滴加溴乙烷(0.8mmol,0.06ml),室温反应,TLC检测反应终点。反应结束后,减压蒸出剩余的卤代烷,加入适量饱和NaCl溶液2mL,再加入乙酸乙酯萃取(4mL×3),合并有机相,无水硫酸镁干燥,过滤,浓缩,真空干燥得白色固体。经硅胶柱色谱纯化,洗脱剂为石油醚/乙酸乙酯=4.5/1(V/V),得白色晶体III267.7mg,产率68.2%。mp120.2~122.6℃;IR(KBr):3200,2924,1728,1458,1384,1262,1180,1159,1042cm-1;1H-NMR(CDCl3,300MHz)δ:5.69(1H,s,N-H,lactam),5.30(t,J=3.5Hz,1H,H-12),4.08~4.05(m,2H,COOCH2CH3),2.67(1H,d,J=8.0Hz,H-18),1.25(t,J=8.7Hz,3H,COOCH2CH3),1.20,1.17,1.13,1.10,0.93,0.90,0.77(21H,7s,CH3×7).ESI-MS:498.4(M+H)+。
一种具有抗肿瘤活性的齐墩果酸化学修饰物及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0