

IPC分类号 : C12N1/21I,C12N15/70I,C12N15/53I,C12N15/54I,C12P13/00I,C12R1/19N





专利摘要
本发明公开了一种基因工程菌及其构建方法及其在生产尼龙12单体12‑氨基月桂酸中的应用,基因工程菌的名称为:EscherichiacoliBL21(DE3):P1‑1‑CGCAB,保藏编号为CCTCCNO:M2019571;构建方法,包括如下内容:一,构建含有重组表达载体A和重组表达载体B的质粒;二,敲除宿主菌的β‑氧化途径关键酶FadD;三,构建重组菌株;通过基因工程的方法解决了菌株中间产物过度氧化的问题,底物的竞争性损失问题,提高了菌株对目标产物的合成效率,能实现胞内辅酶及辅底物自给自足;最终使得基因工程菌具有高效转化月桂酸生产尼龙12单体的能力。
权利要求
1.一种基因工程菌,其特征在于,名称为:Escherichia coli BL21(DE3):P1-1-CGCAB,保藏编号为CCTCC NO:M2019571。
2.一种基因工程菌在生产尼龙12单体12-氨基月桂酸中的应用,其特征在于,名称为:Escherichia coli BL21(DE3):P1-1-CGCAB,保藏编号为CCTCC NO:M2019571;
重组菌株P1-1-CGCAB转化月桂酸生产12-氨基月桂酸的过程为:
将重组菌株P1-1-CGCAB接于TB培养基,温度22-26℃,利用诱导剂进行诱导,诱导时添加促进剂、营养添加剂,诱导11-14小时后收菌;用pH为7.0-8.5的反应缓冲液配置菌泥,添加月桂酸,25-35℃反应,反应8小时后,加入乙腈,离心取上清得12-氨基月桂酸。
说明书
技术领域
本发明涉及基因工程和菌株培养领域,特别是一种基因工程菌及其构建方法及其在生产尼龙12单体12-氨基月桂酸中的应用。
背景技术
尼龙12具有密度小、分解温度高、耐低温性能优良、防噪音效果好等优点,应用广泛。尼龙12可由ω-氨基月桂酸或ω-十二内酰胺单体聚合得到,目前其工业生产主要是利用化学法合成ω-十二内酰胺然后进行开环聚合。以丁二烯为原料合成ω-十二内酰胺,需要经过三聚、催化加氢、氧化、酮化、肟化、贝克曼重排等多个步骤,存在流程长、工艺要求高、产物提取困难、收率低等问题。此外,由于丁二烯来自于石油的C4馏分,其供应受石油市场波动影响较大,而且在合成过程中要使用苯、发烟硫酸等毒性、腐蚀性较大的原料,同时产生大量废弃物,对环境造成巨大的压力。近年来,为了部分填补长碳链尼龙的国内生产空白,我国研究人员开发了发酵法生产十二烷二酸用于合成尼龙1212(刘民英等,2002),但是其反应归根结底是双二酰与双二胺的聚合,除发酵过程外还需要经过三个化学合成步骤才能获得这些单体,并且其发酵原料为石油中的正十二烷,其供应同样受到石油市场的影响。考虑到石油原料的不可再生性和石油市场的复杂性,如果能够开发合成生物学方法利用可再生原料合成长碳链尼龙单体,将具有重大的意义。关于尼龙12单体的生物合成,目前公开发表的资料只有德国多特蒙德工业大学Bühler课题组和韩国建国大学Yun课题组分别于2013年2月、2016年7月和2018年4月发表的研究论文。Bühler课题组首先在大肠杆菌中表达了烷烃单加氧酶AlkBGT和ω-转氨酶CV2025,初步实现了12-氨基月桂酸甲酯的生物合成(Schrewe et al.,2013),然后又在此基础上进一步表达了醇脱氢酶AlkJ、外膜蛋白AlkL和枯草芽孢杆菌来源的丙氨酸脱氢酶AlaDH2,对12-氨基月桂酸甲酯生物合成途径进行了优化,最终全细胞催化3.2mM月桂酸甲酯合成12-氨基月桂酸甲酯的产率为12%(Ladkau etal.,2016)。Yun课题组的最新研究则将12-氨基月桂酸生物合成途径拆分为两个部分,在大肠杆菌中分别表达P450酶(副瘰疬分枝杆菌来源的CYP153A)和负责电子传递的CamA、CamB,以及醇脱氢酶AlkJ和ω-转氨酶mll1207ω-TA,最终利用这两种重组细胞一锅法催化2mM月桂酸,合成12-氨基月桂酸的产率为30%(Ahsan et al.,2018)。在以上两个课题组的研究中,均未解决中间产物12-羰基月桂酸(甲酯)过度氧化生成副产物十二烷二酸的问题,以及人工途径引入后造成胞内辅因子不平衡的问题,这些关键问题将对目标产物的合成效率及其分离纯化造成不良影响。本发明解决以上问题,本发明结合代谢工程和酶工程手段,成功构建了12-氨基月桂酸生物合成途径,并进行了初步优化。
发明内容
为解决现有技术的不足,本发明的目的在于提供一种基因工程菌及其构建方法及其在生产尼龙12单体12-氨基月桂酸中的应用,通过基因工程的方法解决了菌株中间产物过度氧化的问题,底物的竞争性损失问题,提高了菌株对目标产物的合成效率,能实现胞内辅酶及辅底物自给自足;最终使得基因工程菌具有高效转化月桂酸生产尼龙12单体的能力。
为了实现上述目标,本发明采用如下的技术方案:
一种基因工程菌,新菌种的名称为:Escherichia coli BL21(DE3):P1-1-CGCAB,保藏编号为CCTCC NO:M2019571。
一种基因工程菌的构建方法,包括如下内容:一,构建重组质粒,载体质粒包括:pETDuet系列,pACYCDuet系列,pRSFDuet系列,pCDFDuet系列质粒;
重组质粒包括:嵌合P450酶CYP153A-NCP基因的载体质粒;嵌合BsADHC257L基因和Cv2025基因的载体质粒;重组质粒A为含有嵌合P450酶CYP153A-NCP基因与葡萄糖脱氢酶GDH1基因的载体质粒;重组质粒B为含有醇脱氢酶突变体BsADHC257L基因,ω-转氨酶Cv-2025基因与L-丙氨酸脱氢酶AlaDH2基因的载体质粒;二,构建重组菌株,将重组质粒导入宿主大肠杆菌BL21(DE3),获得重组菌株,保存。
前述的一种基因工程菌的构建方法,包括如下内容:构建重组质粒;从M.aquaeolei(DSM 11845)的基因组中利用设计的引物PCR得CYP153A序列,从B.megaterium(KCCM 11745)的基因组中利用设计的引物PCR得P450 BM3氧化还原酶域NCP;利用重叠延伸PCR将CYP153A与NCP融合成CYP153A-NCP,并连入表达载体pETDuet-1中,构建得到质粒pET-T7-CYP153A-NCP;从Chromobacterium violaceum(DSM 30191)的基因组中利用设计的引物PCR得Cv2025的序列,将其连入载体pCD-T7-BsADHC257L,构建得重组质粒:pCD-T7-Cv2025-T7-BsADHC257L;二,构建重组菌株,将质粒pET-T7-CYP153A-NCP与pCD-T7-Cv2025-T7-BsADHC257L,导入大肠杆菌BL21(DE3),获得重组菌株:
BL-CCB:BL21(DE3)(pET-T7-CYP153A-NCP+pCD-T7-Cv2025-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。
前述的一种基因工程菌的构建方法,包括如下内容:一,构建重组质粒,从M.aquaeolei(DSM 11845)的基因组中利用设计的引物PCR得CYP153A序列,从B.megaterium(KCCM 11745)的基因组中利用设计的引物PCR得P450 BM3氧化还原酶域NCP;利用重叠延伸PCR将CYP153A与NCP融合成CYP153A-NCP,并连入表达载体pETDuet-1中,构建得到质粒pET-T7-CYP153A-NCP;从Chromobacterium violaceum(DSM 30191)的基因组中利用设计引物PCR得Cv2025的序列,将其连入载体pCD-T7-BsADHC257L,构建得重组质粒pCD-T7-Cv2025-T7-BsADHC257L;从Bacillus Subtilisin str.168的基因组中利用设计的引物PCR得的序列AlaDH2,将其连入载体pCD-T7-Cv2025-T7-BsADHC257L,构建得重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L;二,构建重组菌株,将质粒pET-T7-CYP153A-NCP与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L,导入大肠杆菌BL21(DE3),获得重组菌株BL-CCAB:BL21(DE3)(pET-T7-CYP153A-NCP+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。
一种基因工程菌的构建方法,包括如下内容:一,构建重组质粒,载体质粒包括:pETDuet系列,pACYCDuet系列,pRSFDuet系列,pCDFDuet系列质粒;重组质粒包括:嵌合P450酶CYP153A-NCP基因的载体质粒;嵌合BsADHC257L基因的载体质粒;嵌合BsADHC257L基因和Cv2025基因的载体质粒;重组质粒A为含有嵌合P450酶CYP153A-NCP基因与葡萄糖脱氢酶GDH1基因的载体质粒;重组质粒B为含有醇脱氢酶突变体BsADHC257L基因,ω-转氨酶Cv-2025基因与L-丙氨酸脱氢酶AlaDH2基因的载体质粒;二,改造宿主大肠杆菌BL21(DE3)的基因背景,改造后的宿主菌包括:改造宿主B-ΔD,改造宿主B1-1,改造宿主P1-1;利用CRISPR/Cas9技术敲除宿主大肠杆菌BL21(DE3)的β-氧化途径关键酶FadD,获得改造宿主B-ΔD;利用CRISPR/Cas9技术在大肠杆菌B-ΔD基因组的原FadD位置敲入了带有LacUV5启动子的AlkL基因,获得改造宿主B1-1;利用CRISPR/Cas9技术在大肠杆菌B1-1基因组的原FadD位置敲入了带有T7启动子的yaaDE基因,获得改造宿主P1-1;三,构建重组菌株,将重组质粒导入改造后的宿主菌,获得重组菌株,保存。
前述的一种基因工程菌的构建方法,包括如下内容:一,构建重组质粒,从M.aquaeolei(DSM 11845)的基因组中利用设计的引物PCR得CYP153A序列,从B.megaterium(KCCM 11745)的基因组中利用设计的引物PCR得P450 BM3氧化还原酶域NCP;利用重叠延伸PCR将CYP153A与NCP融合成CYP153A-NCP,并连入表达载体pETDuet-1中,构建得到质粒pET-T7-CYP153A-NCP;从Chromobacterium violaceum(DSM 30191)的基因组中利用设计引物PCR得Cv2025的序列,将其连入载体pCD-T7-BsADHC257L,构建得重组质粒pCD-T7-Cv2025-T7-BsADHC257L;从Bacillus Subtilisin str.168的基因组中利用设计的引物PCR得的序列AlaDH2,将其连入载体pCD-T7-Cv2025-T7-BsADHC257L,构建得重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L;二,改造宿主大肠杆菌BL21(DE3)的基因背景,利用CRISPR/Cas9技术敲除宿主大肠杆菌BL21(DE3)的β-氧化途径关键酶FadD,获得改造宿主B-ΔD;三,构建重组菌株,将质粒pET-T7-CYP153A-NCP与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L,导入大肠杆菌B-ΔD,获得重组菌B-ΔD-CCAB:BL21(DE3)ΔfadD(pET-T7-CYP153A-NCP+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。
前述的一种基因工程菌的构建方法,包括如下内容:一,构建重组质粒,从M.aquaeolei(DSM 11845)的基因组中利用设计的引物PCR得CYP153A序列,从B.megaterium(KCCM 11745)的基因组中利用设计的引物PCR得P450 BM3氧化还原酶域NCP;利用重叠延伸PCR将CYP153A与NCP融合成CYP153A-NCP,并连入表达载体pETDuet-1中,构建得到质粒pET-T7-CYP153A-NCP;从Chromobacterium violaceum(DSM 30191)的基因组中利用设计引物PCR得Cv2025的序列,将其连入载体pCD-T7-BsADHC257L,构建得重组质粒pCD-T7-Cv2025-T7-BsADHC257L;从Bacillus Subtilisin str.168的基因组中利用设计的引物PCR得的序列AlaDH2,将其连入载体pCD-T7-Cv2025-T7-BsADHC257L,构建得重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L;二,改造宿主大肠杆菌BL21(DE3)的基因背景,利用CRISPR/Cas9技术敲除宿主大肠杆菌BL21(DE3)的β-氧化途径关键酶FadD,获得改造宿主B-ΔD。引入恶臭假单胞菌烷烃降解操纵子中的外膜蛋白AlkL,利用CRISPR/Cas9技术在大肠杆菌B-ΔD基因组的原FadD位置敲入了带有LacUV5启动子的AlkL基因,获得改造宿主B1-1;三,构建重组菌株,将质粒pET-T7-CYP153A-NCP与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L导入大肠杆菌B1-1,获得重组菌B1-1-CCAB:BL21(DE3)ΔfadD::PLacUV5-alkL(pET-T7-CYP153A-NCP+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。
前述的一种基因工程菌的构建方法,包括如下内容:一,构建重组表达载体A和重组表达载体B,从M.aquaeolei(DSM 11845)的基因组中利用设计的引物PCR得CYP153A序列,从B.megaterium(KCCM 11745)的基因组中利用设计的引物PCR得P450 BM3氧化还原酶域NCP;利用重叠延伸PCR将CYP153A与NCP融合成CYP153A-NCP,并连入表达载体pETDuet-1中,构建得到质粒pET-T7-CYP153A-NCP;以构建的质粒pET-T7-CYP153A-NCP为模板,利用设计的引物PCR得CYP153A-NCP-RBS序列;以质粒pET30a-GDH1为模板,利用设计的引物PCR得GDH1-RBS序列;利用重叠延伸PCR将CYP153A-NCP-RBS序列与GDH1-RBS序列融合,并连入表达载体pETDuet-1中,构建得到重组质粒A:pET-T7-CYP153A-NCP-RBS-GDH1;从Chromobacterium violaceum(DSM 30191)的基因组中利用设计引物PCR得Cv2025的序列,将其连入载体pCD-T7-BsADHC257L,构建得重组质粒pCD-T7-Cv2025-T7-BsADHC257L;从Bacillus Subtilisin str.168的基因组中利用设计的引物PCR得的序列AlaDH2,将其连入载体pCD-T7-Cv2025-T7-BsADHC257L,构建得重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L;二,改造宿主大肠杆菌BL21(DE3)的基因背景,利用CRISPR/Cas9技术敲除宿主大肠杆菌BL21(DE3)的β-氧化途径关键酶FadD,获得改造宿主B-ΔD;引入恶臭假单胞菌烷烃降解操纵子中的外膜蛋白AlkL,利用CRISPR/Cas9技术在大肠杆菌B-ΔD基因组的原FadD位置敲入了带有LacUV5启动子的AlkL基因,获得改造宿主B1-1;三,构建重组菌株,将质粒A:pET-T7-CYP153A-NCP-RBS-GDH1与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L导入大肠杆菌B1-1,获得重组菌B1-1-CGCAB:BL21(DE3)ΔfadD::PLacUV5-alkL(pET-T7-CYP153A-NCP-RBS-GDH1+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。
前述的一种基因工程菌的构建方法,包括如下内容:一,构建重组表达载体A和重组表达载体B,从M.aquaeolei(DSM 11845)的基因组中利用设计的引物PCR得CYP153A序列,从B.megaterium(KCCM 11745)的基因组中利用设计的引物PCR得P450 BM3氧化还原酶域NCP;利用重叠延伸PCR将CYP153A与NCP融合成CYP153A-NCP,并连入表达载体pETDuet-1中,构建得到质粒pET-T7-CYP153A-NCP;以构建的质粒pET-T7-CYP153A-NCP为模板,利用设计的引物PCR得CYP153A-NCP-RBS序列;以质粒pET30a-GDH1为模板,利用设计的引物PCR得GDH1-RBS序列;利用重叠延伸PCR将CYP153A-NCP-RBS序列与GDH1-RBS序列融合,并连入表达载体pETDuet-1中,构建得到重组质粒A:pET-T7-CYP153A-NCP-RBS-GDH1;从Chromobacterium violaceum(DSM 30191)的基因组中利用设计引物PCR得Cv2025的序列,将其连入载体pCD-T7-BsADHC257L,构建得重组质粒pCD-T7-Cv2025-T7-BsADHC257L;从Bacillus Subtilisin str.168的基因组中利用设计的引物PCR得的序列AlaDH2,将其连入载体pCD-T7-Cv2025-T7-BsADHC257L,构建得重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L;二,改造宿主大肠杆菌BL21(DE3)的基因背景,利用CRISPR/Cas9技术敲除宿主大肠杆菌BL21(DE3)的β-氧化途径关键酶FadD,获得改造宿主B-ΔD;引入恶臭假单胞菌烷烃降解操纵子中的外膜蛋白AlkL,利用CRISPR/Cas9技术在大肠杆菌B-ΔD基因组的原FadD位置敲入了带有LacUV5启动子的AlkL基因,获得改造宿主B1-1;引入磷酸吡哆醛合成途径基因yaaD和yaaE,利用CRISPR/Cas9技术在大肠杆菌B1-1基因组的原FadD位置敲入了带有T7启动子的yaaDE基因,获得改造宿主P1-1;三,构建重组菌株,将质粒A:pET-T7-CYP153A-NCP-RBS-GDH1与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L导入大肠杆菌P1-1,获得重组菌P1-1-CGCAB:BL21(DE3)ΔfadD::PLacUV5-alkL-PT7-yaaDE(pET-T7-CYP153A-NCP-RBS-GDH1+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。
一种基因工程菌在生产尼龙12单体12-氨基月桂酸中的应用,新菌种的名称为:Escherichia coli BL21(DE3):P1-1-CGCAB,保藏编号为CCTCC NO:M2019571;重组菌株P1-1-CGCAB转化月桂酸生产12-氨基月桂酸的过程为:将重组菌株P1-1-CGCAB接于TB培养基,温度22-26℃,利用诱导剂进行诱导,诱导时添加促进剂、营养添加剂,诱导11-14小时后收菌;用pH为7.0-8.5的反应缓冲液配置菌泥,添加月桂酸,25-35℃反应,反应8小时后,加入乙腈,离心取上清得12-氨基月桂酸。
本发明的有益之处在于:通过选择性引入未检测到有过度氧化问题的P450单加氧酶融合蛋白CYP153A-NCP 和醇脱氢酶突变体BsADHC257L,很好地解决了中间产物过度氧化的问题;针对底物的竞争性损失问题,对底盘细胞进行了代谢改造,通过敲除FadD阻断了β-氧化途径;为了提高底物的跨膜转运效率,表达了异源外膜转运蛋白AlkL,增加了细胞对疏水性底物的摄取;针对各反应步骤之间辅因子不平衡的问题,通过酶库筛选和匹配,精心设计辅酶循环和辅底物循环,通过共表达NADP+葡萄糖脱氢酶GDH1与NAD+依赖的L-丙氨酸脱氢酶AlaDH2,实现了辅因子NADPH与NADH及辅底物L-丙氨酸的胞内自偿;通过在基因工程菌的基因组中插入表达了转氨酶所需辅酶磷酸吡哆醛PLP合成基因yaaDE,实现了胞内辅酶PLP的自给自足。重组菌株P1-1-CGCAB在底物月桂酸浓度为5.0mM时,催化合成12-氨基月桂酸(ADA)的8小时产率达到95.6%以上。
附图说明
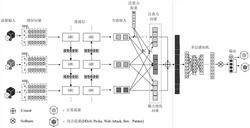
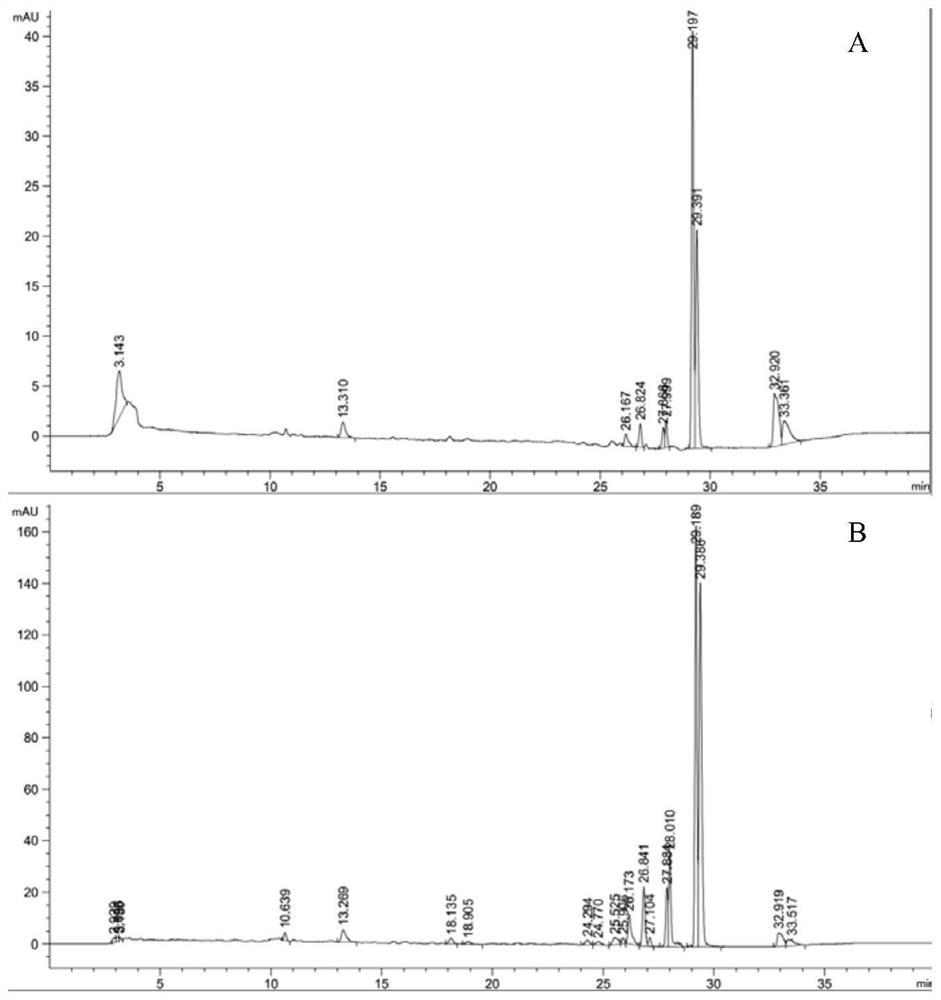
图1是本发明构建方法的一种实施例的流程图;图2为重组菌株BL-CCB破胞粗酶液催化2.5mM月桂酸合成12-氨基月桂酸(ADA)液相图,其中用箭头指出12-氨基月桂酸标准品出峰、合成途径酶催化反应产物出峰、对照出峰;图3中a为过度氧化产物十二烷二酸(DDA)标准品出峰(保留时间为12.893min);b为菌株BL-CCB破胞粗酶液催化2.5mM月桂酸合成12-氨基月桂酸液相图,ADA为产物12-氨基月桂酸出峰(保留时间为10.344min),HAD为中间产物12-羟基月桂酸出峰(保留时间为13.954min);图4为重组菌株BL-CCB利用丙氨酸为氨基供体催化2.5mM月桂酸进行12-氨基月桂酸生物合成,随着合成时间的浓度变化示意图,其中ADA为12-氨基月桂酸,HDA为12-羟基月桂酸;图5为重组菌株BL-CCAB完成辅底物丙氨酸循环与辅酶NADH循环改造后,利用无机铵为氨基供体催化2.5mM月桂酸进行12-氨基月桂酸生物合成,随着合成时间的浓度变化示意图,其中ADA为12-氨基月桂酸,HDA为12-羟基月桂酸;图6为重组菌株B-ΔD-CCAB阻断β-氧化途径后,利用无机铵为氨基供体催化2.5mM月桂酸进行12-氨基月桂酸生物合成,随着合成时间的浓度变化示意图,其中ADA为12-氨基月桂酸,HDA为12-羟基月桂酸;图7为敲入了AlkL的重组菌株B1-1-CCAB利用无机铵为氨基供体催化2.5mM月桂酸进行12-氨基月桂酸生物合成,随着合成时间的浓度变化示意图,其中ADA为12-氨基月桂酸,HDA为12-羟基月桂酸;图8为重组菌株B1-1-CCAB利用无机铵为氨基供体催化5.0mM月桂酸进行12-氨基月桂酸生物合成,随着合成时间的浓度变化示意图,其中ADA为12-氨基月桂酸,HDA为12-羟基月桂酸;图9为重组菌株B1-1-CGCAB利用无机铵为氨基供体催化5.0mM月桂酸进行12-氨基月桂酸生物合成,随着合成时间的浓度变化示意图,其中ADA为12-氨基月桂酸,HDA为12-羟基月桂酸;图10为重组菌株P1-1-CGCAB利用无机铵为氨基供体催化5.0mM月桂酸进行12-氨基月桂酸生物合成,随着合成时间的浓度变化示意图,其中ADA为12-氨基月桂酸,HDA为12-羟基月桂酸;图11为以辅因子及辅底物自给自足的重组菌多酶级联催化利用月桂酸(DA)为底物生产12-氨基月桂酸(ADA)的示意图。
具体实施方式
以下结合附图和具体实施例对本发明作具体的介绍。一种基因工程菌,新菌种的名称为:Escherichia coli BL21(DE3):P1-1-CGCAB,保藏编号为CCTCC NO:M2019571;保藏机构为:中国典型培养物保藏中心(CCTCC);保藏地点为中国武汉;保存日期为:2019年7月19日。如图1所示的一种基因工程菌的构建方法,包括如下内容:一,构建重组质粒,载体质粒包括:pETDuet系列,pACYCDuet系列,pRSFDuet系列,pCDFDuet系列质粒;重组质粒包括:嵌合P450酶CYP153A-NCP基因的载体质粒;嵌合BsADHC257L基因和Cv2025基因的载体质粒;重组质粒A为含有嵌合P450酶CYP153A-NCP基因与葡萄糖脱氢酶GDH1基因的载体质粒;重组质粒B为含有醇脱氢酶突变体BsADHC257L基因,ω-转氨酶Cv-2025基因与L-丙氨酸脱氢酶AlaDH2基因的载体质粒;表达CYP153A-NCP的氨基酸序列为SEQ ID NO:8所示的氨基酸序列或其突变体;SEQ ID NO:8。表达葡萄糖脱氢酶GDH1的氨基酸序列为SEQ ID NO:13所示的氨基酸序列或其突变体,SEQ ID NO:13。表达醇脱氢酶突变体BsADHC257L的氨基酸序列为SEQID NO:9所示的氨基酸序列或其突变体,SEQ ID NO:9。表达ω-转氨酶Cv-2025的氨基酸序列为SEQ ID NO:10所示的氨基酸序列或其突变体,SEQ ID NO:10。表达L-丙氨酸脱氢酶AlaDH2的氨基酸序列为SEQ ID NO:11所示的氨基酸序列或其突变体;SEQ ID NO:11。二,改造宿主大肠杆菌BL21(DE3)的基因背景,改造后的宿主菌包括:改造宿主B-ΔD,改造宿主B1-1,改造宿主P1-1;利用CRISPR/Cas9技术敲除宿主大肠杆菌BL21(DE3)的β-氧化途径关键酶FadD,获得改造宿主B-ΔD;利用CRISPR/Cas9技术在大肠杆菌B-ΔD基因组的原FadD位置敲入了带有LacUV5启动子的AlkL基因,获得改造宿主B1-1;利用CRISPR/Cas9技术在大肠杆菌B1-1基因组的原FadD位置敲入了带有T7启动子的yaaDE基因,获得改造宿主P1-1;需要说明的是:宿主菌包括:BL21(DE3)系列、JM101系列、JM109系列;除了BL21(DE3),其他系列也可以使用,不受限制。在敲除宿主菌的β-氧化途径关键酶FadD后,再在原FadD位置敲入带有LacUV5启动子的源自恶臭假单胞菌烷烃降解操纵子中的外膜蛋白AlkL。这样可以提高大肠杆菌对月桂酸的摄取能力。表达外膜蛋白AlkL的氨基酸序列为SEQ ID NO:12所示的氨基酸序列或其突变体;SEQ ID NO:12。在敲除宿主菌的β-氧化途径关键酶FadD,再在原FadD位置敲入了带有T7启动子的磷酸吡哆醛(PLP)合成基因yaaDE。需要说明的是:核糖-5-磷酸途径依赖的磷酸吡哆醛(PLP)合成基因yaaDE来源没有特别限制,但是优选使用来源于(例如)芽孢杆菌属(Bacillus spp.)等原核生物的基因。表达磷酸吡哆醛(PLP)合成基因yaaD的氨基酸序列为SEQ ID NO:14所示的氨基酸序列或其突变体;SEQ ID NO:14。表达磷酸吡哆醛(PLP)合成基因yaaE的氨基酸序列为SEQ ID NO:15所示的氨基酸序列或其突变体,SEQ IDNO:15。三,构建重组菌株,将含有重组质粒导入宿主菌,获得重组菌株,保存。根据得到的重组菌株全细胞催化月桂酸的反应液中制备12-氨基月桂酸,具体包括以下步骤:将基因工程菌接于TB培养基,温度22-26℃,利用0.05-0.15mM终浓度的IPTG进行诱导,诱导时添加5-氨基乙酰丙酸、维生素B1与微量金属元素,诱导11-14小时后收菌。将菌泥用pH为7.0-8.5的反应缓冲液配成一定菌浓,添加2.0-5.0mM月桂酸,25-35℃反应。反应8小时后,加入等体积乙腈,离心取上清得12-氨基月桂酸。IPTG作为一种诱导剂,5-氨基乙酰丙酸作为一种促进剂,维生素B1与微量金属元素作为营养添加剂。以下用具体的实施例演示产12-氨基月桂酸菌株的构建过程:a)重组质粒的构建;从M.aquaeolei(DSM 11845)的基因组中利用设计引物(上游引物:CYP153A-XbaI-F:
CCTCTAGAAATAATTTTGTTTAACTTTAACAGGAGGAACGGCATGCCGACTTTACCGCGTAC;下游引物:CYP153A-R:
GCTGCCGCCGCTGCCGCCGCTGCCGCCGCTATTCGGGGTCAGTTTCACC)PCR得CYP153A序列,从B.megaterium(KCCM 11745)的基因组中利用设计引物(上游引物:NCP-F:;下游引物:NCP-XhoI-R:)PCR得P450 BM3氧化还原酶域NCP。利用重叠延伸PCR将CYP153A与NCP融合成CYP153A-NCP,并连入表达载体pETDuet-1中,构建得到质粒pET-T7-CYP153A-NCP。CYP153A-NCP核苷酸序列如SEQ ID NO:1。以构建的质粒pET-T7-CYP153A-NCP为模板,利用设计引物(上游引物:CYP153A-XbaI-F:
CCTCTAGAAATAATTTTGTTTAACTTTAACAGGAGGAACGGCATGCCGACTTTACCGCGTAC;下游引物:NCP-RBS-R:
TATATATCTCCTTAGAATTCTTACCCAGCCCACACGTCTTTTG)PCR得CYP153A-NCP-RBS序列。以实验室原有质粒pET30a-GDH1为模板,利用设计引物(上游引物:GDH1-RBS-F:GAATTCTAAGGAGATATATAATGGGTTACAGCGATCTGGAAGG;下游引物:GDH1-XhoI-R:ACTGCTCGAGTTAACCACGACCGGCCTGGAAG)PCR得GDH1-RBS序列。利用重叠延伸PCR将CYP153A-NCP-RBS序列与GDH1-RBS序列融合,并连入表达载体pETDuet-1中,构建得到重组质粒A:pET-T7-CYP153A-NCP-RBS-GDH1。GDH1核苷酸序列如SEQ ID NO:6。从NCBI中获得BsADHC257L(GenBank:KR611715.1)序列信息,其核苷酸序列如SEQ ID NO:2。交由安徽通用公司合成后获得质粒pET30a-BsADHC257L,以其为模板利用设计引物(上游引物:BS-KpnI-F:CGTCGGTACCATGGCAAGCTGGAGCCATCCG;下游引物:BS-XhoI-R:
CAGACTCGAGTTATTTATCTTCCAGGGTCAG)PCR得BsADHC257L的序列,将其连入载体pCDFDuet-1,构建的重组质粒pCD-T7-BsADHC257L。利用设计引物(上游引物:BS-C257L-F:;下游引物:BS-C257L-R:)从Chromobacterium violaceum(DSM 30191)的基因组中利用设计引物(上游引物:CV-BamHI-F:GCCAGGATCCGATGCAAAAACAACGCACCACCTC;下游引物:CV-EcoRI-R:GATCGAATTCTTACGCCAGGCCACGAGCT)PCR得Cv2025的序列,将其连入载体pCD-T7-BsADHC257L,构建得重组质粒pCD-T7-Cv2025-T7-BsADHC257L。Cv2025核苷酸序列如SEQ IDNO:3。从Bacillus Subtilisin str.168的基因组中利用设计引物(上游引物:AlaDH2-EcoRI-RBS-F:GCCAGGATCCGATGCAAAAACAACGCACCACCTC;下游引物:AlaDH2-NotI-R:
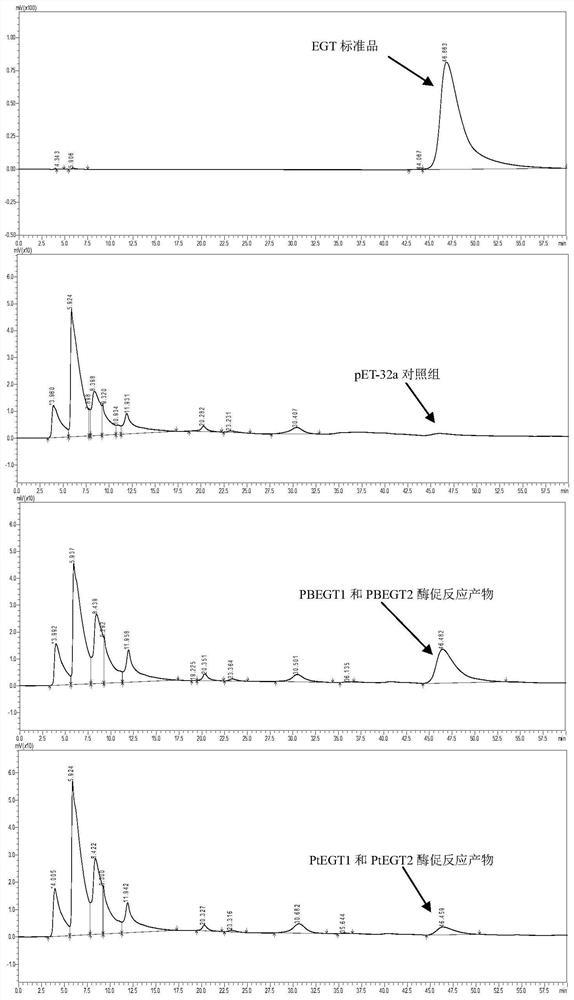
AAGCATTATGCGGCCGCTTAAGCACCCGCCACAGATGATTC)PCR得的序列AlaDH2,将其连入载体pCD-T7-Cv2025-T7-BsADHC257L,构建得重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L。AlaDH2核苷酸序列如SEQ ID NO:4。b)宿主大肠杆菌BL21(DE3)的基因背景改造;为了防止底物被宿主内部脂肪酸代谢途径消耗,利用CRISPR/Cas9技术敲除宿主大肠杆菌BL21(DE3)的β-氧化途径关键酶FadD,获得改造宿主B-ΔD。进一步的,为了加强宿主对疏水性底物月桂酸的摄取,引入恶臭假单胞菌烷烃降解操纵子中的外膜蛋白AlkL。利用CRISPR/Cas9技术在大肠杆菌B-ΔD基因组的原FadD位置敲入了带有LacUV5启动子的AlkL基因,获得改造宿主B1-1。AlkL核苷酸序列如SEQ ID NO:5进一步的,磷酸吡哆醛PLP作为转氨酶和脱羧酶的辅酶,其充足供应有利于转氨反应和脱羧反应的进行。引入枯草芽孢杆菌来源的磷酸吡哆醛合成途径基因yaaD和yaaE,促进底盘细胞内磷酸吡哆醛的再生,为转氨反应提供充足的辅酶,进而提高12-氨基月桂酸的合成效率。利用CRISPR/Cas9技术在大肠杆菌B1-1基因组的原FadD位置敲入了带有T7启动子的yaaDE基因,获得改造宿主P1-1。yaaDE核苷酸序列如SEQ ID NO:7。c)重组菌株的构建;实施例1,将质粒pET-T7-CYP153A-NCP与pCD-T7-Cv2025-T7-BsADHC257L,以常规方法导入大肠杆菌BL21(DE3),获得重组菌株BL-CCB:BL21(DE3)(pET-T7-CYP153A-NCP+pCD-T7-Cv2025-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。实施例2,将质粒pET-T7-CYP153A-NCP与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L,以常规方法导入大肠杆菌BL21(DE3),获得重组菌株BL-CCAB:BL21(DE3)(pET-T7-CYP153A-NCP+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。实施例3,将质粒pET-T7-CYP153A-NCP与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L,以常规方法导入大肠杆菌B-ΔD,获得重组菌B-ΔD-CCAB:BL21(DE3)ΔfadD(pET-T7-CYP153A-NCP+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。实施例4,将质粒pET-T7-CYP153A-NCP与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L以常规方法导入大肠杆菌B1-1,获得重组菌B1-1-CCAB:BL21(DE3)ΔfadD::PLacUV5-alkL(pET-T7-CYP153A-NCP+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。实施例5,将质粒A:pET-T7-CYP153A-NCP-RBS-GDH1与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L以常规方法导入大肠杆菌B1-1,获得重组菌B1-1-CGCAB:BL21(DE3)ΔfadD::PLacUV5-alkL(pET-T7-CYP153A-NCP-RBS-GDH1+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。实施例6,将质粒A:pET-T7-CYP153A-NCP-RBS-GDH1与重组质粒B:pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L以常规方法导入大肠杆菌P1-1,获得重组菌P1-1-CGCAB:BL21(DE3)ΔfadD::PLacUV5-alkL-PT7-yaaDE(pET-T7-CYP153A-NCP-RBS-GDH1+pCD-T7-Cv2025-RBS-AlaDH2-T7-BsADHC257L),并以甘油菌或冻干菌种形式保存。将6个实施例的基因工程菌转化月桂酸生产12-氨基月桂酸,进行产率验证实验:a)重组菌株BL-CCB转化月桂酸生产12-氨基月桂酸;挑取单菌落于5ml含100μg/ml氨苄青霉素与50μg/ml链霉素的LB液体培养基中,37℃、220rpm过夜活化菌种。活化的菌种按2%接种量转接到50ml含相应抗生素(100μg/ml氨苄青霉素,50μg/ml链霉素)的TB液体发酵培养基中,37℃、200rpm培养至OD600约为0.6。加入IPTG至终浓度0.1mM,5-氨基乙酰丙酸0.25mM,0.2mM维生素B1,微量金属元素添加剂(0.5mg MgCl2,30mgFeCl2.6H2O,1mg ZnCl2.4H2O,0.2mg CoCl2.6H2O,1mg Na2MoO4.2H2O,0.5mg CaCl2.2H2O,1mg CuCl2和0.2mg H2BO3),22-26℃,220rpm诱导培养12h后4000xg离心收集菌体细胞。为了验证CYP153A-NCP、BsADHC257L和CV2025是否可以将月桂酸进行催化产生12-氨基月桂酸,将收集菌体用0.9%的氯化钠溶液洗涤两遍后,用转化反应液(100mM Na2HPO4-NaH2PO4缓冲液,pH=8.0,1%葡萄糖,50mM L-丙氨酸)重悬到细胞浓度为50g(湿重)/L,超声破胞,离心取上清粗酶液。2mL粗酶液加月桂酸至终浓度为2.5mM(DMSO终浓度2%),30℃,220rpm转化8小时后,加等体积乙腈终止反应,12000xg离心后,检测上清中12-氨基月桂酸是含量。反应8小时后成功获得了1.0mM终产物ω-氨基月桂酸(图2)。并且,产物中没有过度氧化产物十二烷二酸(DDA)的产生,说明通过选择性引入未检测到有过度氧化问题的P450单加氧酶融合蛋白CYP153A-NCP和醇脱氢酶突变体BsADHC257L,很好地解决了中间产物过度氧化的问题(图3)。另外将收集菌体用0.9%的氯化钠溶液洗涤两遍后,用转化反应液(100mM Na2HPO4-NaH2PO4缓冲液,pH=8.0,1%葡萄糖,50mM L-丙氨酸)重悬到细胞浓度为50g(湿重)/L。全细胞催化2.5mM(DMSO终浓度2%),30℃,220rpm转化,取样加等体积乙腈终止反应,12000xg离心后,检测上清中12-氨基月桂酸的含量。12-氨基月桂酸含量使用反相高效液相色谱检测。HPLC分析使用agilent 1100infinity system,色谱柱为luna C8(2)反向柱(250mm×4.6mm×5μm)。HPLC条件为:流动相A:0.1%TFA水,流动相B:0.1%TFA甲醇。采用梯度洗脱,条件如下:初始:70%A;3min:70%A;20min:15%A;28min:2%A,流速:0.8ml/min;柱温:40±1℃;进样量:25μl。检测使用ELSD检测器(检测温度:65℃,N2流速:1.5ml/min)。检测得反应20h后,12-氨基月桂酸(ADA)的产率达到62%(图4)。b)重组菌株BL-CCAB转化月桂酸生产12-氨基月桂酸;在12-转氨酶催化12-羰基月桂酸生成12-氨基月桂酸的转氨反应中,需要利用丙氨酸作为氨基供体。为了降低成本,在细胞中过表达枯草芽孢杆菌来源的丙氨酸脱氢酶,用于催化胞内的丙酮酸生成丙氨酸,从而实现丙氨酸的再生。同时,由于这个酶是NADH依赖型的,而催化12-羟基月桂酸氧化反应的醇脱氢酶BsADHC257L则是NAD+依赖型的,如此就可以构成途径内部的辅酶NADH循环,而不至于破坏细胞自身的辅酶平衡。养菌过程见构建过程的步骤(a)。将收集菌体用0.9%的氯化钠溶液洗涤两遍后,用转化反应液(100mMNa2HPO4-NaH2PO4缓冲液,pH=8.0,1%葡萄糖,200mM NH3/NH4Cl(NH3:NH4Cl=1:10))重悬到细胞浓度为50g(湿重)/L。全细胞催化2.5mM(DMSO终浓度2%),30℃,220rpm转化后,取样加等体积乙腈终止反应,12000xg离心后,检测上清中12-氨基月桂酸的含量。12-氨基月桂酸含量检测方法见构建过程的步骤(a)。通过导入NADH依赖的AlaDH2与NAD+依赖的BsADHC257L组合实现了辅因子NADH的循环再生,及与L-Ala为辅底物的ω-转氨酶Cv-2025组合实现利用无机铵NH4+循环再生辅底物L-Ala((图11))。检测得反应16小时,12-氨基月桂酸的产率达到65.6%(图5),对比与只能利用大量外加L-Ala来实现转氨反应的重组菌株BL-CCB,重组菌株BL-CCAB由于利用AlaDH2构建了NADH与L-Ala的再生循环,利用无机铵反应更快的达到了62%以上的转化率(图4)。c)重组菌株B-ΔD-CCAB转化月桂酸生产12-氨基月桂酸;由于大肠杆菌中β-氧化途径的关键酶FadD会催化月桂酸形成脂酰辅酶A,使得底物进入β-氧化途径,从而与目标途径竞争底物,因此我们在上述构建的产ω-氨基月桂酸重组大肠杆菌菌株BL-CCAB中敲除了FadD,以减少底物月桂酸的损失。养菌过程见构建过程的步骤(a)。全细胞反应步骤见实施例2-b。12-氨基月桂酸含量检测方法见构建过程的步骤(a)。最后,构建的重组菌株B-ΔD-CCAB以200mM NH3.H2O/NH4Cl(1:10)为氨基供体,以2.5mM月桂酸为底物,18小时12-氨基月桂酸的产率达到76.6%(图6)。对比未敲除FadD的重组菌株BL-CCAB,由于敲除FadD阻断了β-氧化途径,重组菌株B-ΔD-CCAB催化生产12-氨基月桂酸的转化率提高了11%。d)重组菌株B1-1-CCAB转化月桂酸生产12-氨基月桂酸;考虑到月桂酸水溶性差,跨膜运输困难,引入源自恶臭假单胞菌烷烃降解操纵子中的外膜蛋白AlkL以提高大肠杆菌对月桂酸的摄取能力。利用CRISPR/Cas9技术在大肠杆菌B-ΔD-CCAB基因组的原FadD位置敲入了带有LacUV5启动子的alkL。养菌过程见构建过程的步骤(a)。全细胞反应步骤见实施例2-b。12-氨基月桂酸含量检测方法见构建过程的步骤(a)。最后插入AlkL以促进底物转运的重组菌株B1-1-CCAB(ΔfadD::PLacUV5-AlkL)以200mM NH3.H2O/NH4Cl(1:10)为氨基供体,全细胞催化2.5mM月桂酸,12-氨基月桂酸的8小时产率达到了94.8%;而相同情况下,未插入AlkL的重组菌株BL-CCAB(BL21(DE3)WT)的8小时产率为61.6%,重组菌株B-ΔD-CCAB(ΔfadD)的8小时产率为68.2%(图7)。进一步将底物浓度提高到5mM,在相同条件下进行全细胞催化,12-氨基月桂酸的24h产率为53.0%(图8)。e)重组菌株B1-1-CGCAB转化月桂酸生产12-氨基月桂酸;为了实现辅酶NADPH的胞内再生,进而进一步提高12-氨基月桂酸的产率,将NADP+依赖的葡萄糖脱氢酶GDH1引入宿主中,得重组菌株B1-1-CGCAB。养菌过程见构建过程的步骤(a)。全细胞反应步骤见实施例2-b。12-氨基月桂酸含量检测方法见构建过程的步骤(a)。引入的NADP+依赖的葡萄糖脱氢酶GDH1与NADPH依赖的CYP153A-NCP;相配合以实现NADPH的循环再生(图11),最终实现辅酶NADPH胞内再生的重组菌株B1-1-CGCAB以200mM NH3.H2O/NH4Cl(1:10)为氨基供体,全细胞催化5.0mM月桂酸,6小时12-氨基月桂酸的产率达到81.0%(图9)。f)重组菌株P1-1-CGCAB转化月桂酸生产12-氨基月桂酸;为实现转氨酶Cv2025的辅酶磷酸吡哆醛PLP的胞内充足供应,在宿主菌中引入枯草芽孢杆菌来源的磷酸吡哆醛合成途径基因yaaD和yaaE,促进底盘细胞内磷酸吡哆醛的积累。利用CRISPR-Cas9系统对产12-氨基月桂酸的大肠杆菌重组菌株B1-1-CGCAB进行基因组编辑,改造的重组菌株P1-1-CGCAB。养菌过程见构建过程的步骤(a)。全细胞反应步骤见实施例2-b。12-氨基月桂酸含量检测方法见构建过程的步骤(a)。最终在基因组中插入yaaDE加强胞内辅酶磷酸吡哆醛合成的重组菌株P1-1-CGCAB以200mM NH3.H2O/NH4Cl(1:10)为氨基供体,全细胞催化5.0mM月桂酸,8小时12-氨基月桂酸的产率达到95.6%(图10),比未插入yaaDE的B1-1-CGCAB转化率提高了14.6%。以上6个实施例结果说明用本发明的构建策略得到的基因工程菌,可有效促进疏水底物月桂酸的摄取,可实现全细胞催化过程中辅酶及辅底物的胞内自给自足,不需外加辅因子,并可高效催化月桂酸合成12-氨基月桂酸。特别是实施例6得到的重组菌P1-1-CGCAB,为最优基因工程菌,可几乎完全转化5.0mM月桂酸,催化合成12-氨基月桂酸的8小时产率达到95.6%以上,远优于现有技术报道。本发明提供了一种有效提高胞内PLP含量的基因构建策略,实现胞内PLP的高效合成,并保障菌体的正常生长;基于人工酶级联设计的底盘组合代谢工程,通过催化模块的设计和匹配实现NADPH、NADH、L-丙氨酸的再生循环;通过异源表达核糖-5-磷酸途径依赖的磷酸吡哆醛(PLP)合成基因yaaDE,以满足转氨酶对辅酶PLP的需求,整个催化过程不需要外源添加辅酶及辅底物,利用GDH1和丙酮酸连接合成途径与中心代谢途径。所得基因工程菌催化活性高,在底物月桂酸浓度为5.0mM时,催化合成12-氨基月桂酸的8h产率达到95.6%以上。以上显示和描述了本发明的基本原理、主要特征和优点。本行业的技术人员应该了解,上述实施例不以任何形式限制本发明,凡采用等同替换或等效变换的方式所获得的技术方案,均落在本发明的保护范围内。
序列表
<110> 浙江大学
<120> 一种基因工程菌及其构建方法及其在生产尼龙12单体12-氨基月桂酸中的应用
<141> 2019-08-13
<160> 27
<170> SIPOSequenceListing 1.0
<210> 1
<211> 3213
<212> DNA
<213> Marinobacter aqua;B. megaterium
<400> 1
atgccgactt taccgcgtac ctttgatgat atccagagcc gtctgattaa cgccaccagc 60
cgtgttgtgc ctatgcagcg tcagatccaa ggtttaaaat tcttaatgag cgcaaaacgt 120
aaaacctttg gtccgcgccg tccgatgccg gagtttgtgg aaaccccgat tccggatgtg 180
aacacactgg ctttagaaga tatcgacgtg agcaatccgt ttctgtaccg tcaaggccag 240
tggcgcgcct attttaaacg tctgcgcgac gaagccccgg ttcattacca gaaaaacagt 300
ccgttcggcc cgttctggag cgttacccgc ttcgaggaca ttttatttgt ggacaagagt 360
cacgatttat ttagcgcaga gccgcagatt attctgggcg acccgccgga aggtctgagc 420
gtggagatgt tcattgctat ggaccctccg aaacacgacg tgcagcgtag cagtgtgcaa 480
ggtgtggttg ccccgaagaa tttaaaagag atggaaggtt taattcgcag ccgtactggt 540
gatgtgctgg attctttacc gaccgataaa ccgtttaatt gggtgccggc cgttagtaaa 600
gagctgactg gtcgcatgct ggcaacactg ctggattttc cgtacgagga acgccacaag 660
ttagtggagt ggagcgatcg catggctggt gcagcaagcg caaccggtgg tgagtttgcc 720
gatgaaaacg ccatgttcga cgacgcagcc gacatggcac gcagttttag ccgtctgtgg 780
cgcgataaag aagcacgtcg tgccgccggc gaagaaccgg gctttgattt aatcagtctg 840
ctgcagagta ataaagaaac caaagattta attaatcgcc cgatggagtt tattggcaac 900
ttaactttac tgatcgtggc tggtaacgat accacccgta acagcatgag cggcggtctg 960
gttgccatga acgaatttcc gcgcgagttc gaaaagctga aggccaaacc ggagctgatt 1020
cctaacatgg tgagcgagat catccgctgg cagacaccgc tggcatatat gcgtcgcatt 1080
gccaaacaag atgtggagct gggcggtcaa accattaaga aaggcgaccg cgtggtgatg 1140
tggtatgcca gcggcaaccg cgatgaacgc aagtttgata acccggacca attcatcatc 1200
gaccgcaaag acgcccgtaa ccacatgagc tttggctatg gcgtgcatcg ctgcatgggc 1260
aaccgtttag cagaactgca actgcgcatt ctgtgggagg agattttaaa acgcttcgac 1320
aatatcgagg tggtggaaga gccggaacgc gttcagagca atttcgtgcg cggttatagc 1380
cgtttaatgg tgaaactgac cccgaatagc ggcggcagcg gcggcagcgg cggcagcatt 1440
ccttcaccta gcactgaaca gtctgctaaa aaagtacgca aaaaggcaga aaacgctcat 1500
aatacgccgc tgcttgtgct atacggttca aatatgggaa cagctgaagg aacggcgcgt 1560
gatttagcag atattgcaat gagcaaagga tttgcaccgc aggtcgcaac gcttgattca 1620
cacgccggaa atcttccgcg cgaaggagct gtattaattg taacggcgtc ttataacggt 1680
catccgcctg ataacgcaaa gcaatttgtc gactggttag accaagcgtc tgctgatgaa 1740
gtaaaaggcg ttcgctactc cgtatttgga tgcggcgata aaaactgggc tactacgtat 1800
caaaaagtgc ctgcttttat cgatgaaacg cttgccgcta aaggggcaga aaacatcgct 1860
gaccgcggtg aagcagatgc aagcgacgac tttgaaggca catatgaaga atggcgtgaa 1920
catatgtgga gtgacgtagc agcctacttt aacctcgaca ttgaaaacag tgaagataat 1980
aaatctactc tttcacttca atttgtcgac agcgccgcgg atatgccgct tgcgaaaatg 2040
cacggtgcgt tttcaacgaa cgtcgtagca agcaaagaac ttcaacagcc aggcagtgca 2100
cgaagcacgc gacatcttga aattgaactt ccaaaagaag cttcttatca agaaggagat 2160
catttaggtg ttattcctcg caactatgaa ggaatagtaa accgtgtaac agcaaggttc 2220
ggcctagatg catcacagca aatccgtctg gaagcagaag aagaaaaatt agctcatttg 2280
ccactcgcta aaacagtatc cgtagaagag cttctgcaat acgtggagct tcaagatcct 2340
gttacgcgca cgcagcttcg cgcaatggct gctaaaacgg tctgcccgcc gcataaagta 2400
gagcttgaag ccttgcttga aaagcaagcc tacaaagaac aagtgctggc aaaacgttta 2460
acaatgcttg aactgcttga aaaatacccg gcgtgtgaaa tgaaattcag cgaatttatc 2520
gcccttctgc caagcatacg cccgcgctat tactcgattt cttcatcacc tcgtgtcgat 2580
gaaaaacaag caagcatcac ggtcagcgtt gtctcaggag aagcgtggag cggatatgga 2640
gaatataaag gaattgcgtc gaactatctt gccgagctgc aagaaggaga tacgattacg 2700
tgctttattt ccacaccgca gtcagaattt acgctgccaa aagaccctga aacgccgctt 2760
atcatggtcg gaccgggaac aggcgtcgcg ccgtttagag gctttgtgca ggcgcgcaaa 2820
cagctaaaag aacaaggaca gtcacttgga gaagcacatt tatacttcgg ctgccgttca 2880
cctcatgaag actatctgta tcaagaagag cttgaaaacg cccaaagcga aggcatcatt 2940
acgcttcata ccgctttttc tcgcatgcca aatcagccga aaacatacgt tcagcacgta 3000
atggaacaag acggcaagaa attgattgaa cttcttgatc aaggagcgca cttctatatt 3060
tgcggagacg gaagccaaat ggcacctgcc gttgaagcaa cgcttatgaa aagctatgct 3120
gacgttcacc aagtgagtga agcagacgct cgcttatggc tgcagcagct agaagaaaaa 3180
ggccgatacg caaaagacgt gtgggctggg taa 3213
<210> 2
<211> 1296
<212> DNA
<213> Bacillus stearothermophilus
<400> 2
atgccgactt taccgcgtac ctttgatgat atccagagcc gtctgattaa cgccaccagc 60
cgtgttgtgc ctatgcagcg tcagatccaa ggtttaaaat tcttaatgag cgcaaaacgt 120
aaaacctttg gtccgcgccg tccgatgccg gagtttgtgg aaaccccgat tccggatgtg 180
aacacactgg ctttagaaga tatcgacgtg agcaatccgt ttctgtaccg tcaaggccag 240
atggcaagct ggagccatcc gcagtttgaa aaaggtgcca aagcagcagt tgttgaacag 300
tttaaagaac cgctgaaaat taaggaagtg gaaaaaccga ccattagtta tggcgaagtt 360
ctggttcgca ttaaggcatg tggtgtttgt cataccgatc tgcatgcagc ccacggtgac 420
tggccggtga aaccgaaact gccgctgatt ccgggtcatg aaggtgttgg cattgttgaa 480
gaagttggtc cgggcgttac ccatctgaaa gtgggcgata gagtgggcat tccgtggctg 540
tatagcgcct gtggtcattg tgattattgt ctgagcggcc aggaaaccct gtgtgaacat 600
cagaaaaatg ccggctatag cgttgatggc ggttatgcag aatattgtcg cgcagcagca 660
gattatgttg ttaaaattcc ggataatctg agttttgaag aagcagcacc gattttctgt 720
gccggtgtga ccacctataa agccctgaaa gttaccggtg ccaaaccggg tgaatgggtt 780
gcaatctatg gtattggtgg cctgggtcat gtggcagttc agtatgcaaa agcaatgggt 840
ctgaatgtgg ttgcagttga tattggtgac gaaaaactgg aactggccaa agaactgggc 900
gcagatctgg ttgttaatcc gctgaaagaa gatgcagcaa aattcatgaa agaaaaagtt 960
ggcggcgtgc atgcagcagt tgtgaccgcc gtgagcaaac cggcatttca gagtgcctat 1020
aatagtattc gtcgtggcgg cgccctggtg ctggtgggtt tacctccgga agaaatgccg 1080
attccgattt ttgataccgt gctgaatggt attaagatta ttggcagtat tgtgggcacc 1140
cgcaaagatc tgcaagaagc cctgcaattt gccgcagaag gtaaagtgaa aaccattatt 1200
gaagttcagc cgctggaaaa aattaatgaa gtttttgatc gcatgctgaa aggtcagatt 1260
aatggtcgtg tggttctgac cctggaagat aaataa 1296
<210> 3
<211> 1380
<212> DNA
<213> Chromobacterium violaceum
<400> 3
atgcaaaaac aacgcaccac ctcacaatgg cgcgaactgg atgccgcaca ccacctgcac 60
ccgtttaccg acaccgcaag cctgaatcag gccggcgccc gtgttatgac ccgcggcgaa 120
ggtgtgtatc tgtgggattc tgagggtaac aaaattatcg acggcatggc tggtctgtgg 180
tgcgttaatg tcggctatgg tcgtaaagat tttgccgaag cggcccgtcg ccaaatggaa 240
gaactgccgt tctacaacac ctttttcaaa accacgcatc cggcggtggt tgaactgagc 300
agcctgctgg cggaagttac gccggccggc tttgatcgtg tgttctatac caattcaggt 360
tcggaaagcg tggatacgat gatccgcatg gttcgtcgct actgggacgt ccagggcaaa 420
ccggaaaaga aaaccctgat cggtcgttgg aacggctatc atggttctac gattggcggt 480
gcaagtctgg gcggtatgaa atacatgcac gaacagggcg atctgccgat tccgggtatg 540
gcgcatatcg aacaaccgtg gtggtacaaa cacggcaaag atatgacccc ggacgaattt 600
ggtgtcgtgg cagctcgctg gctggaagaa aaaattctgg aaatcggcgc cgataaagtg 660
gcggcctttg ttggcgaacc gattcagggt gcgggcggtg tgattgttcc gccggccacc 720
tattggccgg aaattgaacg tatctgccgc aaatacgatg ttctgctggt cgcagacgaa 780
gttatttgtg gctttggtcg taccggcgaa tggttcggtc atcagcactt tggcttccaa 840
ccggacctgt ttacggcagc taaaggcctg agttccggtt atctgccgat cggcgccgtc 900
ttcgtgggta aacgcgttgc agaaggtctg attgctggcg gtgattttaa tcatggcttc 960
acctatagcg gtcacccggt ctgtgcggcc gtggcacatg ctaatgtggc agctctgcgt 1020
gacgaaggca tcgtgcagcg cgttaaagat gacattggtc cgtatatgca aaaacgttgg 1080
cgcgaaacgt ttagccgttt cgaacacgtc gatgacgtgc gcggcgttgg tatggtccag 1140
gcatttaccc tggtgaaaaa taaagctaaa cgcgaactgt ttccggattt cggcgaaatt 1200
ggtacgctgt gccgtgacat ctttttccgc aacaatctga ttatgcgtgc gtgtggtgat 1260
cacattgtta gcgccccgcc gctggttatg acccgcgcag aagtcgacga aatgctggcc 1320
gtggcggaac gctgcctgga agaatttgaa cagaccctga aagctcgtgg cctggcgtaa 1380
<210> 4
<211> 969
<212> DNA
<213> Bacillus Subtilisin
<400> 4
atggaaaccc tgattctgac ccaggaagaa gttgaaagcc tgattagcat ggatgaagcc 60
atgaatgccg tggaagaagc ctttcgtctg tatgcactgg gtaaagccca gatgccgccg 120
aaagtgtatc tggaatttga aaaaggtgac ctgcgtgcca tgccggccca tctgatgggt 180
tatgcaggcc tgaaatgggt gaatagtcat ccgggcaatc cggataaagg cctgccgacc 240
gtgatggccc tgatgattct gaatagtccg gaaaccggtt ttccgctggc cgtgatggat 300
gccacctata ccaccagtct gcgtaccggt gcagcaggtg gcattgcagc aaaatatctg 360
gcacgtaaaa atagcagcgt gtttggcttt attggctgtg gcacccaggc atattttcag
一种基因工程菌及其构建方法及其在生产尼龙12单体12-氨基月桂酸中的应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0