








专利摘要
本发明提供一种N‑环丙基硝酮化合物的高效催化不对称制备方法,属于有机合成化学技术领域。该方法在反应容器中加入有机溶剂,然后依次加入铜催化剂、手性配体、碱和肟,再加入环丙烯类化合物反应,得到N‑环丙基硝酮化合物。本发明的反应条件温和,由于采用手性配体为双齿膦配体,促进了反应的进行,配体配位的手性金属催化剂造成了非常紧凑的底物结合口袋,有利于实现高对映选择性和非对映选择性控制。同时本发明所用的催化剂和原料肟廉价易得,反应机理新颖,反应的官能团普适性好,为目前较难制备的酮硝酮的合成提供了一条简洁高效的途径,本发明的制备方法得到的产物可以作为治疗癌症、神经退行性疾病等的重要生理活性化合物。
权利要求
1.一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,该方法包括:
在反应容器中加入有机溶剂,然后依次加入铜催化剂、手性配体、碱和肟,再加入环丙烯类化合物反应,得到N-环丙基硝酮化合物。
2.根据权利要求1所述的一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,所述的手性配体为双齿膦配体,结构如L1或L2所示:
3.根据权利要求1所述的一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,所述的肟为二苯甲酮肟、4,4’-二甲基二苯甲酮肟、苯乙酮肟、苯丙酮肟、4-苯基苯甲醛肟或4-甲氧基苯甲醛肟。
4.根据权利要求1所述的一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,所述的环丙烯类化合物为3-甲基-3-苯基环丙烯、3-甲基-3-(2-萘基)环丙烯、3-甲基-3-(4-三氟甲基)苯基环丙烯、3-乙基-3-苯基环丙烯、3-甲基-3-(2-苯乙基)苯基环丙烯、3,3-二苯基环丙烯或3-苯基-3-异丙基环丙烯。
5.根据权利要求1所述的一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,所述的有机溶剂为甲苯、四氢呋喃、二氧六环、乙醚、正己烷、环己烷或乙腈。
6.根据权利要求1所述的一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,所述的铜催化剂为CuCl、CuBr、CuI、Cu(OAc)2、CuOAc、Cu(OTf)2或Cu(CH3CN)4PF6。
7.根据权利要求1所述的一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,所述的碱为叔丁醇钠、叔丁醇锂、叔丁醇钾、苯酚钾或苯酚钠。
8.根据权利要求1所述的一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,所述的反应温度为-50-40℃,反应时间为40-180min。
9.根据权利要求1所述的一种N-环丙基硝酮化合物的高效催化不对称制备方法,其特征在于,所述的环丙烯类化合物、肟和手性配体的摩尔比为1:1.2:(0.25-0.3)。
说明书
技术领域
本发明属于有机合成化学技术领域,具体涉及一种N-环丙基硝酮化合物的高效催化不对称制备方法。
背景技术
硝酮是一类重要的官能团,在有机合成领域、自由基化学、聚合物生产、生物医药领域具有极其重要的应用价值。例如,近二三十年来发展的多种多样的硝酮化合物参与的催化不对称1,3-偶极环加成反应为各种具有重要生理活性的杂环化合物提供了高效的制备方法,也在天然产物合成中有重要应用。(Chem.Rev.1998,98,863-909;Tetrahedron 2007,63(50),12247-12275;Tetrahedron 2007,63,3235-3285;Chem.Rev 2008,108,2887-2902;Chem.Rev.2015,115,5366-5412.)硝酮可以作为自旋捕获试剂(Redox Biol 2016,8,422-9)和生物正交探针(Nanotechnology Reviews 2013,2(2);Curr Opin Chem Biol 2014,21,81-8;Molecules 2015,20,6959-69;Org Biomol Chem 2016,14,7622-38.),在自由基化学和医学检测领域有重要应用价值。在高分子化学领域,硝酮可以用以调控自由基聚合反应。另外,硝酮还可以直接作为治疗一些重大疾病,如肿瘤、抗神经退行性疾病等的药物(Free Radic Biol Med 2008,45(10),1361-74;Future Med Chem 2012,4,1171-1207;Free Radic Biol Med 2013,62,145-56;Biochim Biophys Acta 2014,1840(2),722-9.)。
目前常规使用的合成硝酮的方法仍然局限于经典方法,如醛、酮与羟胺的缩合、胺类、亚胺、羟胺等的氧化、肟的烷基化反应等。这些反应具有很多局限性,如酮硝酮的制备一直是个难题,氧化反应原子经济性和选择性很差等等。重要的是,这些反应的方式大都无法进行催化不对称合成。虽然近期在硝酮合成方面出现了一些新策略,但是一个通用和高效的适合催化不对称反应的方法学仍然没有出现,因此高对映选择性的硝酮合成方法学还未见报道。这些局限性极大地限制了硝酮,尤其是手性硝酮化合物在各个领域的重要应用。
发明内容
本发明的目的是为了解决现有的制备硝酮化合物的方法都无法进行催化不对称合成的问题,而提供一种N-环丙基硝酮化合物的高效催化不对称制备方法。
本发明提供一种N-环丙基硝酮化合物的高效催化不对称制备方法,该方法包括:
在反应容器中加入有机溶剂,然后依次加入铜催化剂、手性配体、碱和肟,再加入环丙烯类化合物反应,得到N-环丙基硝酮化合物。
优选的是,所述的手性配体为双齿膦配体,结构如L1或L2所示:
优选的是,所述的肟为二苯甲酮肟、4,4’-二甲基二苯甲酮肟、苯乙酮肟、苯丙酮肟、4-苯基苯甲醛肟或4-甲氧基苯甲醛肟。
优选的是,所述的环丙烯类化合物为3-甲基-3-苯基环丙烯、3-甲基-3-(2-萘基)环丙烯、3-甲基-3-(4-三氟甲基)苯基环丙烯、3-乙基-3-苯基环丙烯或3-甲基-3-(2-苯乙基)苯基环丙烯、3,3-二苯基环丙烯或3-苯基-3-异丙基环丙烯。
优选的是,所述的有机溶剂为甲苯、四氢呋喃、二氧六环、乙醚、正己烷、环己烷或乙腈。
优选的是,所述的铜催化剂为CuCl、CuBr、CuI、Cu(OAc)2、CuOAc、Cu(OTf)2或Cu(CH3CN)4PF6。
优选的是,所述的碱为叔丁醇钠、叔丁醇锂、叔丁醇钾、苯酚钾或苯酚钠。
优选的是,所述的反应温度为-50-40℃,反应时间为40-180min。
优选的是,所述的环丙烯类化合物、肟和手性配体的摩尔比为1:1.2:(0.25-0.3)。
本发明的有益效果
本发明提供一种N-环丙基硝酮化合物的高效催化不对称制备方法,该方法是在反应容器中加入有机溶剂,然后依次加入铜催化剂、手性配体、碱和肟,再加入环丙烯类化合物反应,得到N-环丙基硝酮化合物。和现有技术相对比,本发明的反应条件温和,由于采用手性配体为双齿膦配体,促进了反应的进行,配体配位的手性金属催化剂造成了非常紧凑的底物结合口袋,有利于实现高对映选择性(高达99%ee)和非对映选择性控制。同时本发明所用的催化剂和原料肟廉价易得,反应机理新颖,反应的官能团普适性好,为目前较难制备的酮硝酮的合成提供了一条简洁高效的途径,本发明的制备方法得到的产物可以作为治疗癌症、神经退行性疾病等的重要生理活性化合物。
附图说明
图1为本发明实施例1制备得到的产物的核磁氢谱图;
图2为外消旋结构的产物的高效液相色谱图;
图3为本发明实施例1制备得到的产物的高效液相色谱图;
图4为本发明实施例2制备得到的产物的核磁氢谱图;
图5为外消旋结构的产物的高效液相色谱图;
图6为本发明实施例2制备得到的产物的高效液相色谱图;
图7为本发明实施例3制备得到的产物的核磁氢谱图;
图8为外消旋结构的产物的高效液相色谱图;
图9为本发明实施例3制备得到的产物的高效液相色谱图;
图10为本发明实施例4制备得到的产物的核磁氢谱图;
图11为外消旋结构的产物的高效液相色谱图;
图12为本发明实施例4制备得到的产物的高效液相色谱图;
图13为本发明实施例5制备得到的产物的核磁氢谱图;
图14为外消旋结构的产物的高效液相色谱图;
图15为本发明实施例5制备得到的产物的高效液相色谱图;
图16为本发明实施例6制备得到的产物的核磁氢谱图;
图17为外消旋结构的产物的高效液相色谱图;
图18为本发明实施例6制备得到的产物的高效液相色谱图;
图19为本发明实施例7制备得到的产物的核磁氢谱图;
图20为外消旋结构的产物的高效液相色谱图;
图21为本发明实施例7制备得到的产物的高效液相色谱图;
图22为本发明实施例8制备得到的产物的核磁氢谱图;
图23为外消旋结构的产物的高效液相色谱图;
图24为本发明实施例8制备得到的产物的高效液相色谱图;
图25为本发明实施例9制备得到的产物的核磁氢谱图;
图26为外消旋结构的产物的高效液相色谱图;
图27为本发明实施例9制备得到的产物的高效液相色谱图;
图28为本发明实施例10制备得到的产物的核磁氢谱图;
图29为外消旋结构的产物的高效液相色谱图。
图30为本发明实施例10制备得到的产物的高效液相色谱图;
图31为本发明实施例2制备得到的产物的结构示意图。
具体实施方式
本发明提供一种N-环丙基硝酮化合物的高效催化不对称制备方法,该方法包括:
在反应容器中加入有机溶剂,然后依次加入铜催化剂、手性配体、碱和肟,再加入环丙烯类化合物反应,得到N-环丙基硝酮化合物。
按照本发明,所述的环丙烯类化合物按照文献(Rubin,et.al,Tetrahedron 2008,64,8610-8617)方法合成。优选为3-甲基-3-苯基环丙烯、3-甲基-3-(2-萘基)环丙烯、3-甲基-3-(4-三氟甲基)苯基环丙烯、3-乙基-3-苯基环丙烯或3-甲基-3-(2-苯乙基)苯基环丙烯、3,3-二苯基环丙烯或3-苯基-3-异丙基环丙烯。
按照本发明,所述的肟是由醛或酮按照现有技术的方法与羟胺缩合而成。优选为二苯甲酮肟、4,4’-二甲基二苯甲酮肟、苯乙酮肟、苯丙酮肟、4-苯基苯甲醛肟或4-甲氧基苯甲醛肟。
按照本发明,所述的手性配体为双齿膦配体,获得方式为商购;本发明由于采用手性配体为双齿膦配体,促进了反应的进行,配体配位的手性金属催化剂造成了非常紧凑的底物结合口袋,有利于实现高对映选择性和非对映选择性控制。所述的双齿膦配体结构如L1或L2所示:
按照本发明,所述的铜催化剂优选一价或二价的铜,更优选为CuCl、CuBr、CuI、Cu(OAc)2、CuOAc、Cu(OTf)2或Cu(CH3CN)4PF6。
按照本发明,所用有机溶剂优选包括甲苯、四氢呋喃、二氧六环、乙醚、正己烷、环己烷或乙腈。
按照本发明,所述的碱包括常用的烷氧基碱金属盐或苯酚碱金属盐,所述烷氧基碱金属盐优选为叔丁醇钠、叔丁醇锂或叔丁醇钾;所述的苯酚碱金属盐优选为苯酚钾或苯酚钠。
按照本发明,所述的反应温度优选为-50-40℃,反应时间优选为40-180min。
按照本发明,所述的环丙烯类化合物、肟和手性配体的摩尔比优选为1:1.2:(0.25-0.3),铜催化剂的加入量优选为环丙烯类化合物加入量的5%;碱的加入量优选为环丙烯类化合物加入量30%。
本发明的制备方法中使用的催化剂铜催化剂和原料肟廉价易得,反应机理十分新颖,通过一系列初步的机理研究表明,该反应经历了一个CuI催化过程,这与涉及铜和肟衍生物的文献报道中涉及单电子转移(SET)过程和自由基过程的报道是完全不同的。C-N键的形成过程经历了一个5并3元环的铜杂retro-Cope氢胺化过渡态。初步的反应机理如下所示:
下面结合具体实施例对本发明作进一步详细的描述,实施例中涉及到的原料均为商购获得。
实施例1
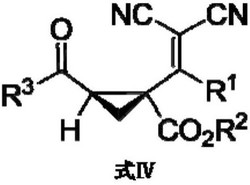
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L1(14.2毫克,0.06毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的二苯甲酮肟(47.3毫克,0.24毫摩尔);将反应体系降温至-50℃,缓慢滴加3-甲基-3-苯基环丙烯(0.2毫摩尔);反应体系放在-50℃温度下搅拌3h,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图1所示)、13C NMR技术、高分辨质谱检测,确定结构为式1,称量重量为62.2毫克,计算产率为95%,图2为外消旋产物的高效液相色谱图,通过图2的外消旋产物的结构确定对照保留时间,然后得到实施例1制备产物的高效液相色谱图(如图3所示),对映选择性为92%;反应可用如下方程式表示:
实施例2
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L1(14.2毫克,0.06毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的二苯甲酮肟(47.3毫克,0.24毫摩尔);将反应体系降温至-50℃,缓慢滴加3-甲基-3-(2-萘基)环丙烯(0.2毫摩尔);反应体系放在-50℃温度下搅拌3h,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图4所示)、13C NMR技术、高分辨质谱检测,确定结构为式2,称量重量为63.4毫克,计算产率为84%,图5为外消旋产物的高效液相色谱图,通过图5的外消旋产物的结构确定对照保留时间,然后得到实施例2制备产物的高效液相色谱图(如图6所示),对映选择性为95%;反应可用如下方程式表示:
本发明实施例2单晶X-射线衍射晶体数据已收入剑桥晶体数据库(CCDC#1548526),结构如图31所示,其晶体结构数据见表1。
表1
实施例3
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L1(14.2毫克,0.06毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的二苯甲酮肟(47.3毫克,0.24毫摩尔);将反应体系降温至-50℃,缓慢滴加3-甲基-3-(4-三氟甲基)苯基环丙烯(0.2毫摩尔);反应体系放在-50℃温度下搅拌3h,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图7所示)、13C NMR技术、高分辨质谱检测,确定结构为式3,称量重量为50.6毫克,计算产率为64%,图8为外消旋产物的高效液相色谱图,通过图8的外消旋产物的结构确定对照保留时间,然后得到实施例3制备产物的高效液相色谱图(如图9所示),对映选择性为96%;反应可用如下方程式表示:
实施例4
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L1(14.2毫克,0.06毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的二苯甲酮肟(47.3毫克,0.24毫摩尔);将反应体系降温至-50℃,缓慢滴加3-乙基-3-苯基环丙烯(0.2毫摩尔);反应体系放在-50℃温度下搅拌3h,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图10所示)、13C NMR技术、高分辨质谱检测,确定结构为式4,称量重量为45.6毫克,计算产率为67%,图11为外消旋产物的高效液相色谱图,通过图11的外消旋产物的结构确定对照保留时间,然后得到实施例4制备产物的高效液相色谱图(如图12所示),对映选择性为99%;反应可用如下方程式表示:
实施例5
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L1(14.2毫克,0.06毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的二苯甲酮肟(47.3毫克,0.24毫摩尔);将反应体系降温至-50℃,缓慢滴加3-甲基-3-(2-苯乙基)苯基环丙烯(0.2毫摩尔);反应体系放在-50℃温度下搅拌3h,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图13所示)、13C NMR技术、高分辨质谱检测,确定结构为式5,称量重量为46.2毫克,计算产率为65%,图14为外消旋产物的高效液相色谱图,通过图14的外消旋产物的结构确定对照保留时间,然后得到实施例5制备产物的高效液相色谱图(如图15所示),对映选择性为96%;反应可用如下方程式表示:
实施例6
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L1(14.2毫克,0.06毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的4,4’-二甲基二苯甲酮肟(54.1毫克,0.24毫摩尔);将反应体系降温至-0℃,缓慢滴加3-甲基-3-苯基环丙烯(0.2毫摩尔);反应体系放在-0℃温度下搅拌60min,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图16所示)、13C NMR技术、高分辨质谱检测,确定结构为式6,称量重量为58.3毫克,计算产率为82%,图17为外消旋产物的高效液相色谱图,通过图17的外消旋产物的结构确定对照保留时间,然后得到实施例6制备产物的高效液相色谱图(如图18所示),对映选择性为90%;反应可用如下方程式表示:
实施例7
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L2(11.7毫克,0.06毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的苯乙酮肟(32.4毫克,0.24毫摩尔);然后缓慢滴加3-甲基-3-苯基环丙烯(0.2毫摩尔);反应体系放在室温度下搅拌40min,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图19所示)、13C NMR技术、高分辨质谱检测,确定结构为式7,称量重量为47.8毫克,计算产率为90%,图20为外消旋产物的高效液相色谱图,通过图20的外消旋产物的结构确定对照保留时间,然后得到实施例7制备产物的高效液相色谱图(如图21所示),对映选择性为94%;反应可用如下方程式表示:
实施例8
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L2(9.7毫克,0.05毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的苯丙酮肟(35.8毫克,0.24毫摩尔);然后缓慢滴加3-甲基-3-苯基环丙烯(0.2毫摩尔);反应体系放在室温度下搅拌40min,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图22所示)、13C NMR技术、高分辨质谱检测,确定结构为式8,称量重量为55.3毫克,计算产率为99%,图23为外消旋产物的高效液相色谱图,通过图23的外消旋产物的结构确定对照保留时间,然后得到实施例8制备产物的高效液相色谱图(如图24所示),对映选择性为97%;反应可用如下方程式表示:
实施例9
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L2(9.7毫克,0.05毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的4-苯基苯甲醛肟(47.3毫克,0.24毫摩尔);然后缓慢滴加3-甲基-3-苯基环丙烯(0.2毫摩尔);反应体系放在室温度下搅拌40min,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图25所示)、13C NMR技术、高分辨质谱检测,确定结构为式9,称量重量为64.8毫克,计算产率为99%,图26为外消旋产物的高效液相色谱图,通过图26的外消旋产物的结构确定对照保留时间,然后得到实施例9制备产物的高效液相色谱图(如图27所示),对映选择性为92%;反应可用如下方程式表示:
实施例10
在25毫升反应管中加入四氢呋喃作为溶剂(2毫升),依次加入氯化亚铜(0.010毫摩尔)、手性配体L2(9.7毫克,0.05毫摩尔)、叔丁醇钠(5.8毫克,0.06毫摩尔)以及底物摩尔质量1.2倍量的4-甲氧基苯甲醛肟(36.3毫克,0.24毫摩尔);然后缓慢滴加3-甲基-3-苯基环丙烯(0.2毫摩尔);反应体系放在室温度下搅拌40min,TLC点板监测,反应完全后,经柱层析分离纯化,得到白色固体,该固体经1H NMR(如图28所示)、13C NMR技术、高分辨质谱检测,确定结构为式10,称量重量为52.9毫克,计算产率为94%,图29为外消旋产物的高效液相色谱图,通过图29的外消旋产物的结构确定对照保留时间,然后得到实施例10制备产物的高效液相色谱图(如图30所示),对映选择性为92%;反应可用如下方程式表示:
一种N-环丙基硝酮化合物的高效催化不对称制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)















动态评分
0.0