

IPC分类号 : H01G11/86,H01G11/44,H01G11/38,C01B31/00










专利摘要
本发明公开了一种超级电容器用微孔炭/石墨烯复合电极材料的制备方法,属于炭材料制备技术领域。该方法是以煤沥青和氧化石墨为碳源,以氢氧化钾为活化剂,将煤沥青和氧化石墨湿法混合,干燥后再与氢氧化钾干法混合,将得到的混合物置于管式炉内,在氮气气氛下进行加热,制得超级电容器用微孔炭/石墨烯复合电极材料。本发明采用氢氧化钾同时活化煤沥青和氧化石墨制备超级电容器用微孔炭/石墨烯复合电极材料的方法,具有制备工艺简单,生产成本低廉,所制备的微孔炭/石墨烯复合电极材料在超级电容器中具有高比容和高倍率性能等优点。
权利要求
1.一种超级电容器用微孔炭/石墨烯复合电极材料的制备方法,其特征在于,该方法具体步骤如下:
(1)反应物的预处理:将氧化石墨溶解于N,N-二甲基甲酰胺中,超声震荡60min后得到混合物A;再将煤沥青溶解于混合物A中,继续超声震荡60min得到混合物B;将混合物B置于磁力搅拌器上加热搅拌直至变为膏状的固体,最后放入鼓风干燥箱中于110℃鼓风干燥后得到混合物C,并研碎;称取一定量氢氧化钾并研碎;将氢氧化钾与混合物C混合研磨得到反应物D;
其中,煤沥青与氧化石墨的质量比介于2/1~11/1之间;氢氧化钾的质量与煤沥青和氧化石墨的总质量的比值为2/1;
(2)微孔炭/石墨烯复合材料的制备:把步骤(1)得到的反应物D放入刚玉瓷舟中,将所述刚玉瓷舟放进管式炉内,以60mL/min的流量通入氮气10min将所述管式炉内的空气排净后,以5℃/min的升温速率加热至800℃,恒温1h后自然降至室温,最后将得到的产物取出、磨碎后放入烧杯中,经酸洗、蒸馏水洗涤和干燥后得到目标产物:超级电容器用微孔炭/石墨烯复合电极材料。
2.如权利要求1所述的一种超级电容器用微孔炭/石墨烯复合电极材料的制备方法,其特征在于,所述步骤(1)中:煤沥青与氧化石墨的质量比为5/1,氢氧化钾与氧化石墨的质量比为12/1。
说明书
技术领域
本发明属于炭材料制备技术领域,具体涉及一种超级电容器用微孔炭/石墨烯复合电极材料的制备方法。
背景技术
超级电容器由于具有功率密度高和使用寿命长等优点而引起人们极大的研究兴趣。但是,超级电容器较低的能量密度限制了其进一步推广应用。超级电容器用电极材料是影响其能量密度的主要因素之一。微孔炭基电极材料由于具有高的比表面积而成为商用超级电容器的主要电极材料之一。但是,微孔炭基电极材料较低的电导率导致微孔炭基超级电容器具有较低的速率性能。如何提高微孔炭基电极材料的电导率并进一步降低其制备成本是当前面临的挑战性课题之一。由于石墨烯材料具有量子霍尔效应、高的理论比表面积(2630m2/g)和高电导率等优点,使得其在储能、催化、传感等众多领域具有潜在的应用前景。
煤沥青,又称煤焦油沥青,是蒸馏煤焦油提取馏分(如轻油、酚油和萘油等)后的副产物,约占煤焦油的50~60%。煤沥青具有含碳量高、原料丰富和价格低廉等优点。以煤沥青为原料,氢氧化钾为活化剂制备的微孔炭具有较高的比表面积和丰富的孔道结构。但是,微孔炭电极材料较差的导电性,降低了其速率性能。在微孔炭电极材料中加入少量的石墨烯可以提高微孔炭/石墨烯电极材料的速率性能。但是,仅通过简单的物理混和制备的微孔炭/石墨烯电极材料,由于微孔炭和石墨烯之间存在较大的接触电阻,导致所得微孔炭/石墨烯电极材料的速率性能不理想。因此,亟需研发一种降低微孔炭/石墨烯复合电极材料内阻的新技术。
中国专利CN102867650A中公开了一种高倍率超级电容器复合电极材料及其制备方法。该方法中,将0.15g氧化石墨和5g葡萄糖溶于水,蒸干水分并研碎所得混合物后,在管式炉中于350℃先预炭化4h,再与5.6g氢氧化钾水溶液混合6h,蒸干所得混合物后,将所得混合物置于管式炉内于800℃恒温活化2h,制得比表面积为2566m2/g的复合材料,在6M KOH溶液中,100mV/s扫速下,复合材料的比容保持在226F/g。中国专利CN103058178A公开了一种高比表面积石墨烯及其制备方法和用途。该方法中,将强碱固体加入氧化石墨烯水溶液中,混匀、干燥去除水分,煅烧后制得多孔石墨烯材料,所得多孔石墨烯的比表面积介于1000~2500m2/g之间。中国专利CN102923698A公开了一种超级电容器用三维多孔石墨烯的制备方法。该方法将氧化石墨烯的水溶液与强碱溶液混匀,预干燥至表面湿润后置于高温下,利用强碱和水蒸气刻蚀石墨烯表面,制得三维多孔石墨烯材料。论文“Synthesis of porous graphene/activated carbon composite with high packing density and large specific surface area for supercapacitor electrode material”(Journal of Power Sources,258(2014)290–296)提出了以氧化石墨、尿素和葡萄糖为碳源,在反应釜中于180℃水热处理24h后得中间产物,然后向中间产物中加入4倍质量的KOH于850℃活化2h后,把所得产物洗净,再向所得产物中加入4倍质量的KOH于850℃再次进行2h的活化,所得复合材料的比表面积达2106m2/g,在6M KOH溶液中,1mV/s扫速下复合材料的比容达到210F/g。论文“Highly conductive and porous activated reduced graphene oxide films for high-power supercapacitors”(Nano Letters,12(2012),1806-1812)将氢氧化钾溶于氧化石墨的水溶液中,其中,氢氧化钾/氧化石墨质量比为14/1,搅拌过滤并干燥滤饼后,在管式炉中于800℃活化,制得产物的比表面积为2400m2/g,在TEABF4/AN有机电解液中,当电流密度为10A/g时,其 比容达到120F/g。
从上述文献中可以看出,所述的多孔石墨烯基电极材料的制备技术中,大多以氧化石墨为单一碳源或者为主要碳源。但是,由于以氧化石墨为碳源制备的多孔石墨烯材料的产率非常低,从而需要制备或购买大量的氧化石墨作碳源。然而,由于采用Hummers法制备氧化石墨的过程复杂,条件苛刻,导致氧化石墨制备成本很高,且相对于煤沥青来说,石墨的价格是高的,从而导致多孔石墨烯基电极材料的价格居高不下。此外,文献中氢氧化钾和氧化石墨的质量的比值较大,即,需要消耗大量的氢氧化钾,这也增加了生产成本,影响了所得材料的推广应用。文件检索结果表明,将煤沥青和氧化石墨二者同时作为碳源,直接制备微孔炭/石墨烯复合电极材料鲜有报道。
发明内容
本发明针对现有微孔炭基复合电极材料制备技术的不足,提出以煤沥青和氧化石墨为碳源,氢氧化钾为活化剂直接法制备超级电容器用微孔炭/石墨烯复合电极材料的方法,以达到简化制备工艺,降低生产成本,提高材料的容量和倍率性能的目的。
本发明具体步骤如下:
(1)反应物的预处理:取一定量的氧化石墨的粉体溶解于DMF(N,N-二甲基甲酰胺)中,超声震荡60min后得到混合物A;再将煤沥青溶解于混合物A中,继续超声震荡60min得到混合物B;将混合物B置于磁力搅拌器上加热搅拌直至液态的混合物B变为膏状的固体,最后将膏状的固体B放入鼓风干燥箱中于110℃鼓风干燥后得到混合物C;称取一定量氢氧化钾并研碎;将氢氧化钾与研碎后的混合物C混合研磨得到反应物D;其中,煤沥青与氧化石墨的质量比介于2/1~11/1之间;氢氧化钾的质量与煤沥青和氧化石墨的总质量的比 值为2/1。
(2)微孔炭/石墨烯复合材料的制备:把步骤(1)得到的反应物D放入刚玉瓷舟中,所述刚玉瓷舟放进管式炉内,以60mL/min的流量通入氮气10min将所述管式炉内的空气排净后,以5℃/min的升温速率加热至800℃,恒温1h后自然降至室温,最后将得到的产物取出、磨碎后放入烧杯中,经酸洗、蒸馏水洗涤和干燥后得到超级电容器用微孔炭/石墨烯复合材料。
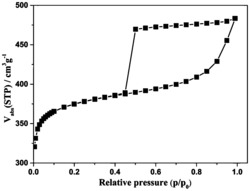
本发明方法是同时以煤沥青和氧化石墨为碳源,以氢氧化钾为活化剂,在氮气保护下进行加热,制得超级电容器用微孔炭/石墨烯复合电极材料。所得复合材料的比表面积介于1749~2233m2/g之间,总孔孔容介于0.82~1.11cm3/g之间,平均孔径介于1.85~2.04nm之间。
作为一种优化,所述步骤(1)中:当煤沥青、氧化石墨、氢氧化钾的质量之比为5:1:12时,所制备的微孔炭/石墨烯复合材料在6M KOH电解液中,在50mA/g电流密度下,电极比容可达278F/g,超级电容器的能量密度可达9.64Wh/kg,在20000mA/g的电流密度下,其比容保持率为78.0%。
本发明的科学原理:
本专利主要利用煤沥青在被加热后具有软化熔融的特点,首先,将煤沥青、氧化石墨和固体氢氧化钾混合均匀后进行加热,在150℃左右,软化熔融后的煤沥青将氧化石墨和氢氧化钾颗粒均匀的包裹起来,最后,在较高温度下,利用氢氧化钾对煤沥青和氧化石墨进行活化,直接合成微孔炭/石墨烯复合电极材料。所得微孔炭/石墨烯复合材料中,微孔炭和石墨烯之间是通过化学键稳定的结合在一起,提高了微孔炭/石墨烯复合电极材料的导电性,从而确保微孔炭/石墨烯复合电极材料具有很好的速率性能。
与现有技术相比,本发明具有以下优点:
1、主要以煤沥青为碳源,以少量的氧化石墨为导电剂,由于煤沥青价廉、易得,减少了微孔炭/石墨烯复合材料的制备成本;
2、采用氢氧化钾同时活化煤沥青和氧化石墨,碱与碳的质量比仅为2/1,可有效降低活化剂的使用量,且该合成方法简单,进一步降低了微孔炭/石墨烯复合材料的制备成本;
3、煤沥青具有线性芳香环取向结构和易于软化熔融的特点,在加热干燥过程能将氧化石墨烯片层连接起来,形成空间立体结构,提高材料的导电性能;
4、在6M KOH电解液中,在50mA/g电流密度下,本发明制备的微孔炭/石墨烯复合材料的比容可达278F/g,在20000mA/g的电流密度下,比容保持率为78.0%。
附图说明
图1是本发明实施例1、2、3、4制备的微孔炭/石墨烯复合材料的氮吸附和脱附等温线。
图2是本发明实施例1、2、3、4制备的微孔炭/石墨烯复合材料的孔径分布图。
图3是本发明实施例2制备的微孔炭/石墨烯复合材料的透射电镜图。
图4是本发明实施例1、2、3、4制备的微孔炭/石墨烯复合材料的比容随电流密度的变化图。
具体实施方式
以下结合具体实施例详述本发明,但本发明不局限于下述实施例。
实施例1
微孔炭/石墨烯复合材料MPC/G11-1-24的具体制备过程如下:
(1)反应物的预处理:取0.5g氧化石墨的粉体溶解于100mL DMF(N, N-二甲基甲酰胺)中,超声震荡60min后得到混合物A;再将5.5g煤沥青溶解于混合物A中,超声震荡60min得到混合物B;将混合物B置于磁力搅拌器上加热搅拌直至液态的混合物B变为膏状固体,最后将膏状固体放入鼓风干燥箱中于110℃鼓风干燥后得混合物C;称取12g氢氧化钾并研碎,将氢氧化钾与混合物C混合研磨得到反应物D。
(2)微孔炭/石墨烯复合材料的制备:把步骤(1)得到的反应物D放入刚玉瓷舟中,所述刚玉瓷舟放进管式炉内,以60mL/min的流量通入氮气10min将所述管式炉内的空气排净后,以5℃/min的升温速率加热至800℃,恒温1h后自然降至室温,最后将得到的产物取出、磨碎后放入烧杯中,经2mol/L盐酸洗涤后,再经蒸馏水洗涤至中性,于110℃干燥24h后,过325目筛,得到超级电容器用微孔炭/石墨烯复合材料。所得微孔炭/石墨烯复合材料标记为MPC/G11-1-24。在6M KOH电解液中,在50mA/g电流密度下,MPC/G11-1-24电极的比容达250F/g,MPC/G11-1-24超级电容器的能量密度达8.69Wh/kg。
实施例2
微孔炭/石墨烯复合材料MPC/G10-2-24的具体制备过程如下:
(1)反应物的预处理:取1g氧化石墨的粉体溶解于200mL DMF(N,N-二甲基甲酰胺)中,超声震荡60min后得到混合物A;再将5g煤沥青溶解于混合物A中,超声震荡60min得到混合物B;随后,按照与实施例1中的步骤(1)同样的方法实施。
(2)按照与实施例1中的步骤(2)同样的方法实施,所得微孔炭/石墨烯复合材料标记为MPC/G10-2-24。在6M KOH电解液中,在50mA/g电流密度下,MPC/G10-2-24电极的比容可达278F/g,MPC/G10-2-24超级电容器的能量密度可达9.64Wh/kg,在20000mA/g的电流密度下,其比容保持率为78.0%。
实施例3
微孔炭/石墨烯复合材料MPC/G9-3-24的具体制备过程如下:
(1)反应物的预处理:取1.5g氧化石墨的粉体溶解于300mL DMF(N,N-二甲基甲酰胺)中,超声震荡60min后得到混合物A;再将4.5g煤沥青溶解于混合物A中,超声震荡60min得到混合物B;随后,按照与实施例1中的步骤(1)同样的方法实施。
(2)按照与实施例1中的步骤(2)同样的方法实施,所得微孔炭/石墨烯复合材料标记MPC/G9-3-24。在6M KOH电解液中,在50mA/g的电流密度下,MPC/G9-3-24电极的比容达275F/g,MPC/G9-3-24超级电容器的能量密度达9.55Wh/kg。
实施例4
微孔炭/石墨烯复合材料MPC/G8-4-24的具体制备过程如下:
(1)反应物的预处理:取2g氧化石墨的粉体溶解于400mL DMF(N,N-二甲基甲酰胺)中,超声震荡60min后得到混合物A;再将4g煤沥青溶解于混合物A中,超声震荡60min得到混合物B;随后,按照与实施例1中的步骤(1)同样的方法实施。
(2)按照与实施例1中的步骤(2)同样的方法实施,所得微孔炭/石墨烯复合材料标记MPC/G8-4-24。在6M KOH电解液中,在50mA/g的电流密度下,MPC/G8-4-24电极的比容达231F/g,MPC/G8-4-24超级电容器的能量密度达8.05Wh/kg。
实施例1-4所得微孔炭/石墨烯复合材料的孔结构分析结果和产率见表1。
表1实施例1-4所得微孔炭/石墨烯复合材料的孔结构分析结果和产率
一种超级电容器用微孔炭/石墨烯复合电极材料的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0