







专利摘要
本发明公开了一种制备托吡酯的方法,该方法将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯溶于有机溶剂中与固体碳酸铵反应,具体步骤如下:在将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯充分溶解于有机溶剂中后加到固体碳酸铵及适量脱水剂的混合物中,反应后浓缩除溶剂,加入适量的乙酸乙酯及活性炭脱色,再过滤除活性炭并浓缩滤液至干得托吡酯粗品,加入异丙醇和正己烷的混合溶剂重结晶托吡酯粗品,冷却结晶后过滤并充分干燥滤饼即可得到托吡酯纯品。本发明用固体碳酸铵代替氨气进行氨解反应,操作简便、安全,降低设备成本,有利于工业化生产。
说明书
技术领域技术领域
本发明属于制药领域,特别是一种癫痫症药物托吡酯的制备方法。
技术背景背景技术
托吡酯(Topiramate)是一个由氨基磺酸酯取代单糖的新型抗癫痫药物,其药理作用为选择性阻断电压依赖的钠通道。托吡酯还可以提高γ-氨基丁酸(GABA),激活GABA受体的频率,从而加强GABA诱导氯离子内流的能力,增强神经系统中抑制性神经递质作用。另外,托吡酯可以降低谷氨酸AMPA受体的活性,从而起到减弱神经系统中兴奋性中枢神经递质的作用。
托吡酯的化学名称为:2,3:4,5-双-O-(1-甲基亚乙基)-β-D-吡喃果糖氨基磺酸酯,其结构式如下:
1985年的美国专利US 4,513,006,1986年的美国专利US 4,582,916及J.Med.Chem.,1987,30(5):880-887中均给出了下面的路线制备托吡酯。
采用双丙酮叉果糖与氨基磺酰氯反应直接得到托吡酯,操作简单。但是,该路线存在两大缺陷:1、要求NaH与DMF化合,这是一个不可控制的放热反应,因此是潜在的炸药;2、国内市场上,氨基磺酰氯难以商业化应用,需要使用高毒性和强腐蚀性的氯磺酰异氰酸酯(CSI)制备。由于其毒性和腐蚀性,氯磺酰异氰酸酯难以使用,且其价格较贵。
1993年的欧洲专利EP 0,533,483公开了另一种制备托吡酯的路线,见下图。
该方法是用2,3:4,5-双-O-(1-甲基亚乙基)-β-D吡喃果糖氯磺酸酯与金属叠氮化合物反应,后经催化氢化将磺酸酯还原成托吡酯。但该路线中使用到了叠氮化合物,在处理叠氮化合物时可能会发生爆炸,安全性不高。另一方面,该路线还包括一附加的化学转化反应,即把叠氮化合物还原成胺基部分,增加了操作的复杂性。
1995年的美国专利US 5,387,700及一些中国专利、世界专利,中国医药工业杂志,1999,30(11):486-487中均提供了另一种途径制备托吡酯,见下图。
该路线中使用了氨气及绝对无水的溶剂进行氨解反应。一方面,工业生产中,气体存在很多不利的因素:如反应装置有比较严格的要求(如:气密性、抗压性等);多余氨气需要回收,这就需要气体回收装置,增加了设备的成本;气体的操作、运输、储存没有液体或固体来的方便、安全。另一方面,采用绝对无水的溶剂,这就要求反应中使用到的溶剂在反应前必须进行除水,增加了成本,不经济且增加了操作的复杂性。
2004年的世界专利WO 078,769也公布了一种托吡酯的合成路线,见下图。
该路线是用2,3:4,5-双-O-(1-甲基亚乙基)-β-D吡喃果糖氯磺酸酯与二氨基磺酸在120~140℃条件下反应得到托吡酯。由于反应需要使用高温条件,增加了工业生产的成本,不适合工业化生产。
2004年的欧洲专利EP 1,627,881公开了托吡酯合成的另一种方法,见下图。
该路线中使用了氯磺酸异氰酸酯(CSI),具有高毒性和强腐蚀性,国内市场且难以商业化使用,故不适宜于工业生产中应用。另一方面,水解时需要高温条件,在一定的程度上增加了生产成本。
2007年的中国专利CN 101,045,740也提供了一种制备托吡酯的方法。该方法是用2,3:4,5-双-O-(1-甲基亚乙基)-β-D吡喃果糖氯磺酸酯与氨水反应制备托吡酯,见下图。
该路线中采用2,3:4,5-双-O-(1-甲基亚乙基)-β-D吡喃果糖氯磺酸酯与氨水反应制备托吡酯,用氨水代替氨气进行氨解反应,操作简便、安全,降低设备成本,且溶剂不需要无水处理,后处理中只萃取就可以得到产物,有利于工业化生产。但是,另一方面,2,3:4,5-双-O-(1-甲基亚乙基)-β-D吡喃果糖氯磺酸酯在碱性条件下极易水解成2,3:4,5-双-O-(1-甲基亚乙基)-β-D吡喃果糖,这样一来,最终得到托吡酯的量很少甚至根本得不到托吡酯。
发明内容发明内容
本发明的目的在于提供一种新的且制备符合药用标准的托吡酯的方法,该方法利用易于得到的固体碳酸铵为原料,在安全的条件下反应,并得到相对较高的产量。该方法避免使用了氨气,使得操作安全简便,经济性和安全性在一定程度上都有所提高。
实现本发明目的的技术解决方案为:一种托吡酯的制备方法,将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯溶于有机溶剂中与固体碳酸铵反应,具体步骤如下:
第一步,将化合物2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯充分溶解于有机溶剂中;
第二步,将第一步所得溶液加入到固体碳酸铵及脱水剂的混合物中进行反应,然后浓缩除溶剂,加入乙酸乙酯及活性炭脱色,再过滤除活性炭并浓缩滤液至干得托吡酯粗品;
第三步,加入异丙醇和正己烷的混合溶剂重结晶托吡酯粗品,冷却结晶后过滤、干燥滤饼即可得到托吡酯纯品。
本发明与现有技术相比,其显著优点:1、用固体碳酸铵代替氨气进行氨解反应所带来的操作简便,安全,降低了设备成本,经济性和安全性都有所提高,利于工业化生产;2、体系采用四氢呋喃和乙腈的混合溶剂,一方面缩短了反应时间,另一方面提高了产物得率;3、反应过程中加入适量的脱水剂,减少了副产物的生成,提高了托吡酯的产量。
附图说明附图说明
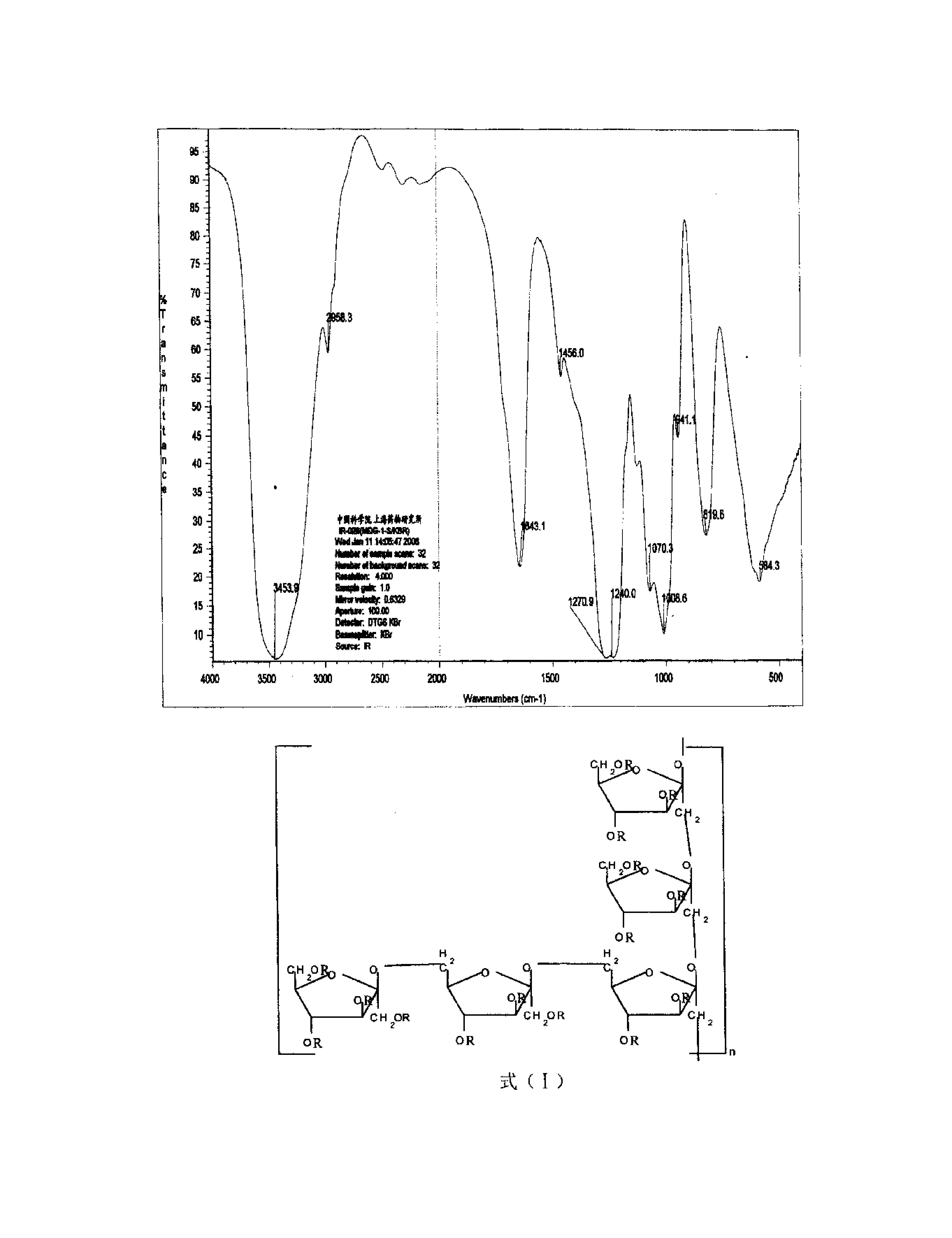
附图是托吡酯的1H-NMR图。
具体实施方式具体实施方式
下面结合附图对本发明作进一步详细描述。
本发明一种托吡酯的制备方法合成托吡酯的路线如下:
本发明一种托吡酯的制备方法,将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯溶于有机溶剂中与固体碳酸铵反应,具体步骤如下:在35~95℃下,将化合物2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯充分溶解于有机溶剂中后加到固体碳酸铵及适量脱水剂的混合物中,反应0.5-24小时后,浓缩除溶剂,加入适量的乙酸乙酯及活性炭脱色,再过滤除活性炭并浓缩滤液至干得托吡酯粗品,加入异丙醇和正己烷的混合溶剂重结晶托吡酯粗品,冷却结晶后过滤并充分干燥滤饼即可得到托吡酯纯品。
所述的有机溶剂为非质子有机溶剂为乙酸乙酯、四氢呋喃、甲苯、乙腈或其中任意几种的混合溶剂。其中,非质子有机溶剂优先为四氢呋喃、乙腈或两者以任意比例的混合溶剂。脱水剂可为无水硫酸钠、无水硫酸镁、无水碳酸钠或无水碳酸钾等。
所述的2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯与固体碳酸铵的摩尔比为1.0~3.0。第二步中加入乙酸乙酯的体积是2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯质量的4倍,加入活性炭的质量是2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯质量的0.2倍。第三步中异丙醇和正己烷的体积比为1∶4。
化合物2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯的制备可参考文献《中国医药工业杂志》,1999,30(11):486-487。
下面结合具体实施例对本发明进行进一步描述。
实施例1:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
65℃下,将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯(5g,13.9mmol)溶于四氢呋喃溶液(50mL),加入固体碳酸铵(2.67g,27.8mmol)及无水硫酸钠(0.79g,5.6mmol),24h后反应完全,浓缩除四氢呋喃,加入20mL乙酸乙酯及1g活性炭脱色。过滤除活性炭,浓缩滤液至干并加入异丙醇和正己烷的混合液重结晶。冷却结晶,过滤并充分干燥滤饼得到白色晶体3.75g,收率为80%。mp122~123℃,1H-NMR(300MHz,CDCl3),δ:5.03(br s,2H,NH2),3.77-4.64(m,7H),1.35,1.43(2s,6,4,5-CH3),1.49,1.56(2s,6,2,3-CH3)。
实施例2:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
35℃下,按照实施例1的操作,收率为68%。
实施例3:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
95℃下,按照实施例1的操作,收率为54%。
实施例4:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
按照实施例1的操作,用乙酸乙酯替换四氢呋喃,收率为33%。
实施例5:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
按照实施例1的操作,用乙醇替换四氢呋喃,收率为0%。
实施例6:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
65℃下,将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯(5g,13.9mmol)溶于的乙腈溶液(50mL),加入固体碳酸铵(2.67g,27.8mmol)及无水硫酸钠(0.79g,5.6mmol),0.5h后反应完全,浓缩除乙腈,加入20mL乙酸乙酯及1g活性炭脱色。过滤除活性炭,浓缩滤液至干并加入异丙醇和正己烷的混合液重结晶。冷却结晶,过滤并充分干燥滤饼得到白色晶体2.4g,收率为51%
实施例7:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
65℃下,将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯(5g,13.9mmol)溶于四氢呋喃和乙腈的混合溶液(50mL,四氢呋喃/乙腈=1/1),加入固体碳酸铵(2.67g,27.8mmol)及无水硫酸钠(0.79g,5.56mmol),2h后反应完全,浓缩除溶剂,加入20mL乙酸乙酯及1g活性炭脱色。过滤除活性炭,浓缩滤液至干并加入异丙醇和正己烷的混合液重结晶。冷却结晶,过滤并充分干燥滤饼得到白色晶体3.64g,收率为77%。
实施例8:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
按照实施例7的操作,不加任何脱水剂,收率为61%。
实施例9:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
按照实施例7的操作,用无水MgSO4替换无水Na2SO4,收率为60%。
实施例10:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
按照实施例7的操作,用无水K2CO3替换无水Na2SO4,收率为40%。
实施例11:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
65℃下,将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯(26.5g,73.9mmol)溶于265mL四氢呋喃溶液中,加入固体碳酸铵(14.22g,14.8mmol)及无水硫酸钠(4.20g,29.6mmol),24h后反应完全,浓缩除溶剂,加入106mL乙酸乙酯及5.3g活性炭脱色。过滤除活性炭,浓缩滤液至干并加入异丙醇和正己烷的混合液重结晶。冷却结晶,过滤并充分干燥滤饼得到白色晶体19.52g,收率为78%。
实施例12:2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氨基磺酸酯的制备
65℃下,将2,3:4,5-双-O-(1-甲基亚乙基)-D-吡喃果糖氯磺酸酯(19.7g,54.9mmol)溶于四氢呋喃和乙腈的混合溶液(197mL,四氢呋喃/乙腈=1/1),加入固体碳酸铵(10.52g,109.5mmol)及无水硫酸钠(3.2g,22.5mmol),4h后反应完全,浓缩除溶剂,加入79mL乙酸乙酯及4g活性炭脱色。过滤除活性炭,浓缩滤液至干并加入异丙醇和正己烷的混合液重结晶。冷却结晶,过滤并充分干燥滤饼得到白色晶体13.89g,收率为75%。
一种托吡酯的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0