









专利摘要
本发明公开了一种氟替卡松类衍生物的关键中间体17-β羧酸的合成方法,所述方法以式(Ⅲ)或式(Ⅳ)所示的化合物为原料,在强碱和次卤酸盐氧化剂的作用下,对应制备得到式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物。本发明所用的氧化剂便宜易得,环境污染小,并且反应选择性好,收率高,后处理简单,产品易分离,适合工业化生产。
权利要求
1.一种式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物的合成方法,其特征在于以式(Ⅲ)或式(Ⅳ)所示的化合物为原料,在强碱和次卤酸盐氧化剂的作用下,对应制备得到式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;
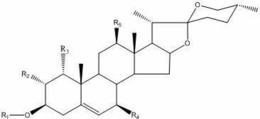
式(I)或式(III)中,R1为H或卤素;R2、R3各自独立为H、OH或卤素;R4为H、OH或C1~C4的直链烷基;
式(II)或式(IV)中,R5为H或卤素,R6为H、OH或C1~C4的直链烷基;
所述卤素为F、Cl、Br或I。
2.如权利要求1所述的方法,其特征在于所述的方法包括如下步骤:将式(Ⅲ)或式(Ⅳ)所示的化合物、次卤酸盐、强碱在反应溶剂中,在-20℃~50℃温度下搅拌反应,反应1~10小时,反应结束后,反应液经后处理,对应制得式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;
所述式(Ⅲ)或式(Ⅳ)所示的化合物、强碱、次卤酸盐的物质的量之比为1.0:10.0~26.0:3.0~9.0;
所述次卤酸盐为次氯酸盐、次溴酸盐或次碘酸盐。
3.如权利要求1或2所述的方法,其特征在于所述式(I)或式(III)中,R1为H或F;R2为F;R3为OH;R4为甲基。
4.如权利要求1或2所述的方法,其特征在于所述式(II)或式(IV)中,R5为H或F;R6为甲基。
5.如权利要求1或2所述的方法,其特征在于所述次卤酸盐为次溴酸钠、次氯酸钠或次碘酸钠。
6.如权利要求1或2所述的方法,其特征在于所述强碱为氢氧化钠或氢氧化钾。
7.如权利要求2所述的方法,其特征在于所述的反应溶剂为水和有机溶剂的混合,所述有机溶剂为下列一种或两种以上的混合:甲醇、乙醇、异丙醇、叔丁醇、乙二醇二甲醚、乙二醇二乙醚、乙醚、丙醚、异丙醚、丁醚、苯甲醚、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、二氯甲烷、乙腈、二甲基甲酰胺。
8.如权利要求2所述的方法,其特征在于所述反应液后处理方法为:反应液中加入过量的还原剂的水溶液,在-20℃~50℃下充分搅拌,然后加酸调pH值到6~7,减压蒸馏回收有机溶剂,剩余液调pH值到2~3,加少量水析出固体,过滤,滤饼烘干,对应制得式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;所述还原剂为亚硫酸氢钠或亚硫酸钠。
9.如权利要求2所述的方法,其特征在于所述的方法包括如下步骤:次卤酸盐和强碱在溶剂水中,在-20℃~50℃温度下,加入式(Ⅲ)或式(Ⅳ)所示的化合物和有机溶剂,-20℃~50℃温度下搅拌反应,反应1~10小时,反应结束后,反应液中加入过量的还原剂亚硫酸氢钠或亚硫酸钠的水溶液,在-20℃~50℃下搅拌15~60分钟,然后加酸调pH值到6~7,减压蒸馏回收有机溶剂,剩余液调pH值到2~3,加少量水析出固体,过滤,滤饼烘干,对应制得式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;所述有机溶剂为下列一种或两种以上的混合:甲醇、乙醇、异丙醇、叔丁醇、乙二醇二甲醚、乙二醇二乙醚、乙醚、丙醚、异丙醚、丁醚、苯甲醚、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、二氯甲烷、乙腈、二甲基甲酰胺。
10.如权利要求8或9所述的方法,其特征在于所述还原剂、次卤酸盐的物质的量之比为2~2.5:1,所述还原剂的水溶液的质量百分浓度为10~15%。
说明书
(一)技术领域
本发明涉及一种丙酸(糠酸)氟替卡松及其类似物关键中间体17-β羧酸(Ⅰ)、(Ⅱ)的合成方法。
(二)背景技术
丙酸氟替卡松(Fluticasone propionate)为德国葛兰素威康公司开发的一种新型糖皮质激素药物,该药物与其他糖皮质激素相比具有很强的疗效和较弱的不良反应,糠酸氟替卡松( fluticasone furoate), 是由葛兰素史克( GSK )公司研发,2007年4月在美国上市的新药鼻喷雾剂,商品名为Veramyst。与丙酸氟替卡松相比,糠酸氟替卡松的受体亲和力更高,药效时间延长。
目前丙酸(糠酸)氟替卡松及其类似物关键中间体17-β羧酸(Ⅰ)、(Ⅱ)一般都是由(Ⅴ)、(Ⅵ)经高碘酸氧化得到(WO2004001369,CN100497367C,WO2008115069),该方法采用的原料合成成本较高;也有文献报道采用空气氧化(Ⅴ)、(Ⅵ)得到(Ⅰ)、(Ⅱ)(WO2004052912,J. Org. Chem., 1986, 51, 2315-2328)但是反应时间长,收率偏低;中国专利(CN1244593C)采用双氧水氧化氟米松21-乙酸酯得到17-β羧酸(Ⅰ);中国专利(CN101838301)报道了从(Ⅲ)、(Ⅳ)经过上碘,置换,水解,氧化得到(Ⅰ)、(Ⅱ),路线繁琐,成本较高,总体收率较低。
(三)发明内容
本发明目的在于提供一种经济合理且工艺稳定的丙酸(糠酸)氟替卡松及其类似物关键中间体17-β羧酸(Ⅰ)、(Ⅱ)的制备方法,采用相对便宜的原料(Ⅲ)、(Ⅳ)在次卤素酸盐的氧化下一步合成(Ⅰ)、(Ⅱ),本发明提供的原料价格便宜,缩短了氟替卡松的合成路线,提高收率和产品质量,降低成本,适合工业化生产。
本发明采用的技术方案是:
一种式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物的合成方法,以式(Ⅲ)或式(Ⅳ)所示的化合物为原料,在强碱和次卤酸盐氧化剂的作用下,对应制备得到式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;
式(I)或式(III)中,R1为H或卤素;R2、R3各自独立为H、OH或卤素;连接R4的单键为R或者S构型,R4为H、OH或C1~C4的直链烷基;
式(II)或式(IV)中,R5为H或卤素,连接R6的单键为R或者S构型,R6为H、OH或C1~C4的直链烷基;
所述卤素为F、Cl、Br或I。
所述连接R4的单键为R或者S构型,是指与R4相连接的碳为手性碳,手性碳连接R4的单键为R或者S构型均可,不影响本发明方法的实施。
所述连接R6的单键为R或者S构型,是指与R6相连接的碳为手性碳,手性碳连接R6的单键为R或者S构型均可,不影响本发明方法的实施。
进一步,所述式(I)或式(III)中,R1优选为H或F;R2优选为F;R3优选为OH;R4优选为甲基。
进一步,所述式(II)或式(IV)中,R5优选为H或F;R6优选为甲基。
进一步,本发明所述的方法包括如下步骤:将式(Ⅲ)或式(Ⅳ)所示的化合物、次卤酸盐、强碱在反应溶剂中,在-20℃~50℃温度下搅拌反应,反应1~10小时,反应结束后,反应液经后处理,对应制得式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;
所述式(Ⅲ)或式(Ⅳ)所示的化合物、强碱、次卤酸盐的物质的量之比为1.0:10.0~26.0:3.0~9.0,优选为1:10~15:3~6
所述次卤酸盐为次氯酸盐、次溴酸盐或次碘酸盐。
进一步,本发明所述次卤酸盐优选为次溴酸钠、次氯酸钠或次碘酸钠,最优选次溴酸钠或次氯酸钠。
所述强碱为氢氧化钠或氢氧化钾,优选氢氧化钠。
本发明所述的反应溶剂为水和有机溶剂的混合,所述有机溶剂为下列一种或两种以上的混合:甲醇、乙醇、异丙醇、叔丁醇、乙二醇二甲醚、乙二醇二乙醚、乙醚、丙醚、异丙醚、丁醚、苯甲醚、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、二氯甲烷、乙腈、二甲基甲酰胺,优选为四氢呋喃或1,4-二氧六环。所述反应溶剂中,所述水的体积用量通常以强碱的质量计为5~20mL/g。所述有机溶剂的体积用量通常以式(Ⅲ)或式(Ⅳ)所示的化合物的质量计为10~30mL/g。
本发明所述反应液后处理方法为:反应液中加入过量的还原剂的水溶液,在-20℃~50℃下充分搅拌(通常搅拌15~60分钟),然后加酸调pH值到6~7,减压蒸馏回收有机溶剂,剩余液调pH值到2~3,加少量水析出固体,过滤,滤饼烘干,对应制得式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;所述还原剂为亚硫酸氢钠或亚硫酸钠。
所述还原剂优选为亚硫酸氢钠。
所述还原剂的用量相对于次卤酸盐过量即可,用于还原未反应完的氧化剂,通常加入的还原剂、次卤酸盐的物质的量之比为2~2.5:1。所述还原剂的水溶液的质量百分浓度通常为10~15%。
所述剩余液调pH值到2~3,加少量水析出固体,是指剩余液调pH值到2~3后,根据其中水的含量多少,已有少量固体析出,可再加少量水,使产物完全析出即可。有时剩余液中含的水较多,加酸调pH值后,可能产物已经完全析出,此时不加水也可以。
更进一步,推荐本发明所述的方法包括如下步骤:次卤酸盐和强碱在溶剂水中,在-20℃~50℃温度下,加入式(Ⅲ)或式(Ⅳ)所示的化合物和有机溶剂,-20℃~50℃温度下搅拌反应,反应1~10小时,反应结束后,反应液中加入过量的还原剂亚硫酸氢钠或亚硫酸钠的水溶液,在-20℃~50℃下搅拌15~60分钟,然后加酸调pH值到6~7,减压蒸馏回收有机溶剂,剩余液调pH值到2~3,加少量水析出固体,过滤,滤饼烘干,对应制得式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;所述有机溶剂为下列一种或两种以上:甲醇、乙醇、异丙醇、叔丁醇、乙二醇二甲醚、乙二醇二乙醚、乙醚、丙醚、异丙醚、丁醚、苯甲醚、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、二氯甲烷、乙腈、二甲基甲酰胺。
本发明所述次卤酸盐中,次氯酸钠可直接配成水溶液,加入反应液中进行反应,所述次溴酸钠和次碘酸钠由于性质不稳定,不能长时间存放,必须在反应时现场配制。常用次溴酸钠的制备方法是:NaOH 溶于水中,降温到-20~-5℃,缓慢滴加溴,滴加时控制反应液温度低于0℃,滴加完毕即制得所述含有次溴酸钠的水溶液,需立即进行下步反应。由于本发明方法中也需要NaOH参与反应,因此制备次溴酸钠时,NaOH的物质的量相对于溴过量,即按照1摩尔NaOH与1摩尔溴反应制得1摩尔次溴酸钠,则参与本发明反应的实际NaOH的用量为NaOH的投料量减去溴的投料量,次溴酸钠的物质的量按照溴的物质的量来计量。
较为优选的,氧化剂次卤酸盐为次溴酸钠时,推荐本发明方法按以下步骤进行:NaOH 溶于水中,降温到-20~-5℃,缓慢滴加溴,滴加时控制反应液温度低于0℃,滴加完毕即制得所述含有次溴酸钠的水溶液,加入式(Ⅲ)或式(Ⅳ)所示的化合物和有机溶剂,-20℃~10℃(优选5~8℃)温度下搅拌反应,反应1~10小时(优选4小时),反应结束后,反应液加入过量的还原剂亚硫酸氢钠或亚硫酸钠的水溶液,在-20℃~10℃(优选5~8℃)温度下搅拌15~60分钟,然后加酸调pH值到6~7,减压蒸馏回收有机溶剂,剩余液调pH值到2~3,加少量水析出固体,过滤,滤饼烘干,对应制得式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;所述式(Ⅲ)或式(Ⅳ)所示的化合物、生成次溴酸钠后剩余的NaOH、次溴酸钠的物质的量之比为1.0:10.0~26.0:3.0~9.0,即相当于式(Ⅲ)或式(Ⅳ)所示的化合物、NaOH总的投料量、溴的物质的量之比为1.0:13.0~35.0:3.0~9.0,优选为1:13~25:3~5,最优选为1:13~20:3.3~5。所述水的体积用量通常以NaOH的质量计为5~20mL/g,优选10mL/g。所述有机溶剂的体积用量通常以式(Ⅲ)或式(Ⅳ)所示的化合物的质量计为10~30mL/g,优选10~20mL/g。所述有机溶剂优选为四氢呋喃或1,4-二氧六环。
较为优选的,氧化剂次卤酸盐为次氯酸钠时,推荐本发明方法按以下步骤进行:NaOH 溶于水中,降温到-20~-5℃,加入式(Ⅲ)或式(Ⅳ)所示的化合物和有机溶剂,缓慢滴加5~15wt%次氯酸钠的水溶液,滴加时控制反应液温度低于0℃,滴加完毕后,-10~30℃(优选5~25℃)温度下搅拌反应,反应1~10小时(优选4小时),反应结束后,反应液加入过量的还原剂亚硫酸氢钠或亚硫酸钠的水溶液,在-10~30℃(优选5~25℃)温度下搅拌15~60分钟,然后加酸调pH值到6~7,减压蒸馏回收有机溶剂,剩余液调pH值到2~3,加少量水析出固体,过滤,滤饼烘干,对应制得式(Ⅰ)或式(Ⅱ)所示的17-β羧酸衍生物;所述式(Ⅲ)或式(Ⅳ)所示的化合物、NaOH、次氯酸钠的物质的量之比为1.0:10.0~26.0:3.0~9.0,优选为1:10~15:3~6,最优选为1:13~15:5~6。所述总的水的体积用量通常以NaOH的质量计为5~20mL/g,优选9~10mL/g。所述有机溶剂的体积用量通常以式(Ⅲ)或式(Ⅳ)所示的化合物的质量计为10~30mL/g,优选10~20mL/g。所述有机溶剂优选为四氢呋喃或1,4-二氧六环。所述总的水的体积用量是用于溶解NaOH的水和次氯酸钠水溶液中溶剂水的总和。
所述加酸调pH值到6~7,一般使用浓硫酸、浓盐酸或浓硝酸,优选浓硫酸。
所述剩余液调pH值到2~3,一般使用浓硫酸来调pH值。
本发明的有益效果在于:(1)所用氧化剂环境污染小,价格便宜,使用的原料(Ⅲ)、(Ⅳ)相对于(Ⅴ)、(Ⅵ)也相对低廉;(2)反应条件温和操作简单,反应时间短;(3)反应选择性好,收率高,后处理简单,产品易分离,合工业化生产。
(四)具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
实施例中,原料9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮 由浙江仙居君业药业有限公司提供。
6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮 按照CN 100429221C中公开的合成方法制备得到。
6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮按照CN 101838301 A中公开的合成方法制备得到。
实施例1~4
实施例1
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和15ml水,搅拌溶解降温到-5℃,缓慢滴加溴1.5g (9.3mmol),控制温度低于0℃,半小时滴加完毕,加入1,4-二氧六环20ml和9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.8mmol)在5℃下搅拌反应4h,反应完毕,加入1.9g NaHSO3和19ml水,搅拌半小时,用浓硫酸调pH值到中性,减压除去二氧六环,用浓硫酸调 pH值到2~3,加入水10ml,白色固体析出,过滤,滤饼干燥得到9β,11β-环氧-17α羟基-16α甲基-孕甾-1,4-二烯-3-酮-17β-羧酸0.8g,收率80%。
1H NMR (400 MHz, CDCl3), δ:0.97 (d, J=7.0Hz, 3H), 1.09 (s, 3H), 1.33-1.40 (m, 1H), 1.45 (s, 3H), 1.49-1.83 (m, 4H), 2.08-2.71 (m, 6H), 2.96 (m, 1H), 3.21 (s, 1H), 6.16 (s, 1H), 6.20 (dd, J1=10.0 Hz, J2=1.6 Hz, 1H), 6.60 (d, J=10.0Hz, 1H); MS(ESI-) m/z 357.2 (M-H)+.
实施例2
在装有温度计的100ml的三口烧瓶中加入NaOH 2.0g (50.0mmol)和20ml水,搅拌溶解降温到-5℃,缓慢滴加溴2.0g (12.4mmol),控制温度低于0℃,半小时滴加完毕,加入1,4-二氧六环20ml和9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.8mmol)在8℃下搅拌反应4h,反应完毕,加入2.6g NaHSO3和26ml水,搅拌半小时,用浓硫酸调pH值到中性,减压除去二氧六环,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3-酮-17β-羧酸0.85g,收率85%。
实施例3
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和5ml水,搅拌溶解降温到-5℃,加入THF 10ml和9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.8mmol),缓慢滴加10%的次氯酸钠10ml (15mmol),控制温度低于0℃,半小时滴加完毕,在5℃下搅拌反应4h,反应完毕,加入3.1g NaHSO3和31ml水,搅拌半小时,用浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3-酮-17β-羧酸0.8g,收率80%。
实施例4
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和5ml水,搅拌溶解降温到-5℃,加入THF 10ml和9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.8mmol),缓慢滴加10%的次氯酸钠10ml (15mmol),控制温度低于0℃,半小时滴加完毕,在25℃下搅拌反应4h,反应完毕,加入3.1g NaHSO3和31ml水,搅拌半小时,用浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3-酮-17β-羧酸0.82g,收率82%。
实施例5~8
实施例5
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和15ml水,搅拌溶解降温到-5℃,缓慢滴加溴1.5g (9.3mmol),控制温度低于0℃,半小时滴加完毕,加入THF 20ml和6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.6mmol)在5℃下搅拌反应4h,反应完毕,加入1.9g NaHSO3和19ml水,搅拌半小时,用浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,加入水10ml,白色固体析出,过滤,滤饼干燥得到6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,-酮-17β-羧酸0.75g,收率75%。
1H NMR (400 MHz, CDCl3), δ:0.97 (d,3H, J=7.0Hz), 1.08 (s, 3H), 1.42 (s, 3H), 1.64 (q, 2H,J =12.0Hz), 1.76–1.85 (m, 2H), 2.15 (d, 2H, J=11.0 Hz),2.32 (m, 2H, ), 2.63–2.70 (m, 1H), 2.94–3.02 (m, 1H), 3.32 (s, 1H), 5.36–5.52 (2m, 1H), 6.25 (dd,1H, J1=10.0Hz, J2=1.6Hz), 6.45 (s, 1H), 6.52 (d, 1H, J=10.0Hz); MS(ESI-) m/z 375.2 (M-H)+.
实施例6
在装有温度计的100ml的三口烧瓶中加入NaOH 2.0g (50.0mmol)和20ml水,搅拌溶解降温到-5℃,缓慢滴加溴2.0g (12.4mmol),控制温度低于0℃,半小时滴加完毕,加入THF 20ml和6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.6mmol)在8℃下搅拌反应4h,反应完毕,加入2.6g NaHSO3和26ml水,搅拌半小时,用浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,-酮-17β-羧酸0.80g,收率80%。
实施例7
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和5ml水,搅拌溶解降温到-5℃,加入THF 20ml和6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g(2.6mmol),缓慢滴加10%的次氯酸钠10ml (15mmol),控制温度低于0℃,半小时滴加完毕,在5℃下搅拌反应4h,反应完毕,加入3.1g NaHSO3和31ml水,搅拌半小时,浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,-酮-17β-羧酸0.73g,收率73%。
实施例8
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和5ml水,搅拌溶解降温到-5℃,加入THF 20ml和6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.6mmol),缓慢滴加10%的次氯酸钠10ml (15mmol),控制温度低于0℃,半小时滴加完毕,在25℃下搅拌反应4h,反应完毕,加入3.1g NaHSO3和31ml水,搅拌半小时,浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到6α-氟-9β,11β-环氧-17α-羟基-16α-甲基-孕甾-1,4-二烯-3,-酮-17β-羧酸0.75g,收率75%。
实施例9~12
实施例9
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和15ml水,搅拌溶解降温到-5℃,缓慢滴加溴1.5g (9.3mmol),控制温度低于0℃,半小时滴加完毕,加入THF 10ml和6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.5mmol)在5℃下搅拌反应4h,反应完毕,加入1.9g NaHSO3和19ml水,搅拌半小时,用浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,加入水10ml,白色固体析出,过滤,滤饼干燥得到6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3-酮-17β-羧酸0.80g,收率80%。
1H NMR (400 MHz, DMSO-d6) δ: 0.86 (d, 3H, J=7.2Hz), 0.99 (s,3H), 1.49, (s, 3H,), 4.42 (d, 1H, J=11.2Hz), 5.54–5.70 (2m, 1H), 6.08 (s, 1H), 6.27 (dd,1H, J1=10.0Hz, J2=1.6Hz), 7.24 (d, 1H, J=10.4Hz), 12.40 (s, 1H); MS(ESI-) m/z 395.2 (M-H)+.
实施例10
在装有温度计的100ml的三口烧瓶中加入NaOH 2.0g (50.0mmol)和20ml水,搅拌溶解降温到-5℃,缓慢滴加溴2.0g (12.4mmol),控制温度低于0℃,半小时滴加完毕,加入1,4-二氧六环20ml和6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g (2.5mmol)在8℃下搅拌反应4h,反应完毕,加入2.6g NaHSO3和26ml水,搅拌半小时,用浓硫酸调pH值到中性,减压除去二氧六环,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3-酮-17β-羧酸0.85g,收率85%。
实施例11
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和5ml水,搅拌溶解降温到-5℃,加入THF 10ml和6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g(2.5mmol),缓慢滴加10%的次氯酸钠10ml (15mmol),控制温度低于0℃,半小时滴加完毕,在5℃下搅拌反应4h,反应完毕,加入3.1g NaHSO3和31ml水,搅拌半小时,浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3-酮-17β-羧酸0.8g,收率80%。
实施例12
在装有温度计的100ml的三口烧瓶中加入NaOH 1.5g (37.5mmol)和5ml水,搅拌溶解降温到-5℃,加入THF 10ml和6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3,20-二酮1.0g(2.5mmol),缓慢滴加10%的次氯酸钠10ml (15mmol),控制温度低于0℃,半小时滴加完毕,在25℃下搅拌反应4h,反应完毕,加入3.1g NaHSO3和31ml水,搅拌半小时,浓硫酸调pH值到中性,减压除去THF,用浓硫酸调pH值到2~3,白色固体析出,过滤,滤饼干燥得到6α,9α-二氟-11β,17α-二羟基-16α-甲基-孕甾-1,4-二烯-3-酮-17β-羧酸0.82g,收率82%。
一种氟替卡松类衍生物的关键中间体17-β羧酸衍生物的合成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0