

IPC分类号 : C07J63/00,C07F15/00,A61P35/00,A61K31/58









专利摘要
本发明公开了三例熊果酸哌嗪二硫代甲酸‑吡啶钌配合物及其制备方法和应用。本发明所述的熊果酸哌嗪二硫代甲酸‑吡啶钌配合物的制备方法为:将配体熊果酸哌嗪二硫代甲酸钠分别与三(2,2'‑联吡啶)二氯化钌、三(1,10‑菲咯啉)二氯化钌或三(4,7‑二苯基‑1,10‑菲咯啉)二氯化钌置于第一有机溶剂中进行配位反应,即得相应的目标物粗品。申请人的试验表明,这些配合物对某些肿瘤细胞株具有极为显著的抑制活性,且大大高于其母核和顺铂,同时其对顺铂耐药株细胞的抑制活性显著优于顺铂,有望用于抗肿瘤药物的制备。
权利要求
1.具有下述式(I)、(II)或(III)所示结构的熊果酸哌嗪二硫代甲酸-吡啶钌配合物:
上述式(I)、(II)或(III)所示配合物的阴离子均为氯离子。
2.权利要求1所述熊果酸哌嗪二硫代甲酸-吡啶钌配合物的制备方法,其特征是,主要包括以下步骤:
取下述式(L)所示配体和下述式(1)、(2)或(3)所示化合物置于第一有机溶剂中,于加热或不加热条件下进行配位反应,制得相应的目标物粗品;
3.根据权利要求2所述的制备方法,其特征是,在反应之前加入脱氢剂。
4.根据权利要求3所述的制备方法,其特征是,所述的脱氢剂为选自四甲基氢氧化铵、吡啶、三乙胺、氢氧化钠、氢氧化钾、碳酸钾和碳酸铯中的一种或两种以上的组合。
5.根据权利要求2~4中任一项所述的制备方法,其特征是,所述的第一有机溶剂为选自甲醇、乙醇、丙醇、叔丁醇、乙二醇甲醚、二氯甲烷和氯仿中的一种或两种以上的组合。
6.根据权利要求2~4中任一项所述的制备方法,其特征是,所述式(L)所示配体按下述方法进行制备:
取下述式(UAP)所示化合物和二硫化碳置于第二有机溶剂中,在碱性条件下进行缩合反应,制得配体粗品;
7.根据权利要求6所述的制备方法,其特征是,在式(L)所示配体的制备方法中,所述的第二有机溶剂为选自甲醇、乙醇、四氢呋喃、二氧六环、二氯甲烷、氯仿和环己烷中的一种或两种以上的组合。
8.根据权利要求2~4中任一项所述的制备方法,其特征是,还包括对制得的目标物粗品进行纯化的步骤。
9.权利要求1所述配合物在制备抗肿瘤药物中的应用。
10.一种药物组合物,含有治疗上有效剂量的权利要求1所述配合物。
说明书
技术领域
本发明涉及涉及医药技术领域,具体涉及熊果酸哌嗪二硫代甲酸-吡啶钌配合物及其制备方法和应用。
背景技术
钌金属基抗肿瘤配合物被认为是除了铂类金属基抗肿瘤配合物以外的最有前途的抗癌药物之一(Mestroni G,Alessio E.Metal Based Drugs,1994,1:41-48),其代表化合物NAMI2A和[ImH][trans2 RuCl4(dmso2S)(Im)](Im=imidazole)具有优良的抗癌活性。由于钌金属基抗肿瘤配合物具有毒性低、易吸收、排泄快和易肿瘤组织吸收等优点(SavaG,Alessio E,Bergamo A,Mestroni G.Topics in Biological Inorganic Chemistry,Vol.1.Berlin:Springer,1999.143-169),新型高效低毒钌类金属基抗肿瘤配合物的设计与筛选受到药物化学家的高度关注。
熊果酸及其衍生物是一类天然三萜羧酸化合物,在诱导肿瘤细胞凋亡、抑制肿瘤细胞侵袭和转移、抑制炎性反应、逆转放化疗药物的耐药性、阻滞细胞周期、诱导细胞自噬、抑制上皮-间充质转化、抑制肿瘤血管生成及调节免疫功能等方面发挥重要的抗肿瘤作用(冯振,陈伟霞,丁亚杰,赵爱光,熊果酸抗肿瘤作用机制的研究进展,癌症进展,2019,17(14),1613-1615,1674)。如公开号为CN102675405A的发明专利,公开了一类含异丙醇胺亚结构的乙酰氧基熊果酸哌嗪类化合物及其制备方法,并指出该类熊果酸哌嗪类化合物在防治农业病虫害方面的用途。又如公开号为CN101891794A的发明专利,公开了一种具有抗肿瘤活性的熊果酸哌嗪类衍生物及其制备方法,但从其体外活性试验可知,其制得的目标化合物仅对人宫颈癌细胞Helae有杀伤作用(《疾病模型与实验病理学》,李才,吉林大学出版社,2002年12月,第49-50页)。但目前尚未见有熊果酸哌嗪二硫代甲酸-吡啶钌配合物及其制备方法以及其对细胞毒性的相关报道。
发明内容
本发明要解决的技术问题是提供一类结构新颖、对某些肿瘤细胞毒性高的熊果酸哌嗪二硫代甲酸-吡啶钌配合物及其制备方法和应用。
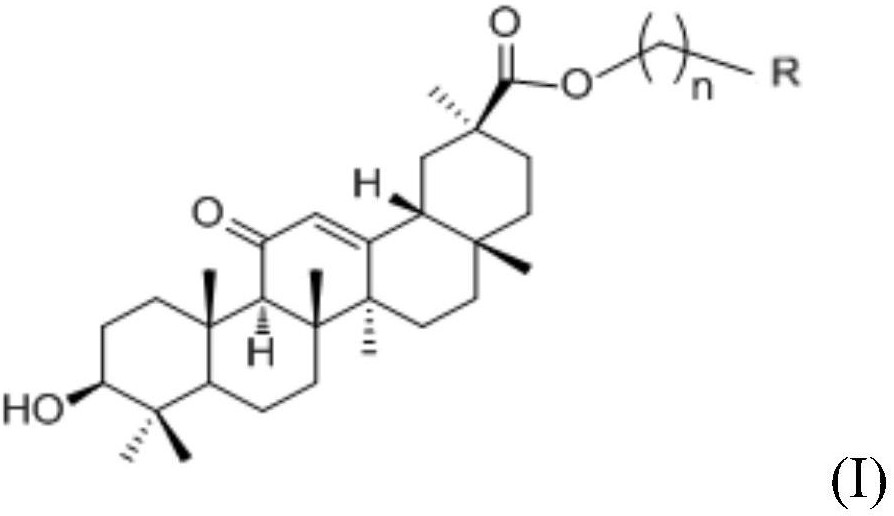
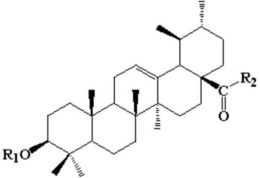
本发明所述的熊果酸哌嗪二硫代甲酸-吡啶钌配合物为具有下述式(I)、(II)或(III)所示结构的化合物或其药学上可接受的盐:
本发明所述熊果酸哌嗪二硫代甲酸-吡啶钌配合物的制备方法,主要包括以下步骤:取下述式(L)所示配体和下述式(1)、(2)或(3)所示化合物置于第一有机溶剂中进行配位反应,制得相应的目标物粗品;
上述配合物的制备方法中,式(L)所示配体和式(1)、(2)或(3)所示化合物的摩尔比为化学计量比,在实际的操作中,在实际的操作中,式(L)所示化合物,或者是式(1)、(2)或(3)所示化合物也可以相对过量,但过量会造成所得产物不纯。
上述配合物的制备方法中,所述的第一有机溶剂为选自甲醇、乙醇、丙醇、叔丁醇、乙二醇甲醚、二氯甲烷和氯仿中的一种或两种以上的组合。所述第一有机溶剂的用量以能够溶解参加反应的原料为宜,通常情况下,以1mmol的式(L)所示化合物为基准,所有参加反应的原料通常用10~30mL的第一有机溶剂来溶解。
上述配合物的制备方法中,通过在反应之前加入脱氢剂可以有效提高整个反应的产率。所述脱氢剂能够溶于水或第一有机溶剂,优选可以是选自四甲基氢氧化铵、吡啶、三乙胺、氢氧化钠、氢氧化钾、碳酸钾和碳酸铯中的一种或两种以上的组合。所述脱氢剂的加入量通常为式(L)所示配体摩尔量的5%以上,优选为15~30%。所述脱氢剂优选用水或第一有机溶剂溶解后再加入到反应体系中。
上述配合物的制备方法中,反应可以在加热或不加热或冰浴条件下进行,相对而言,反应在加热条件进行时能够获得更高的产率。优选反应在≥40℃的条件下进行,更优选是在50~70℃条件下进行。反应过程中采用TLC跟踪监测反应是否完全。根据申请人的经验,当反应在50~70℃条件下进行时,反应时间控制在24~48h较为适宜。
上述配合物的制备方法制得的是目标物的粗品,可采用现有常规的纯化方法对其进行纯化以提高目标配合物的纯度。在本申请中,优选采用中性氧化铝柱色谱法过柱分离来进行纯化,洗脱时以二氯甲烷和甲醇的组合物为冼脱剂,优选以二氯甲烷和甲醇按100~50:2的体积比组成的组合物为冼脱剂。在洗脱剂的组成中,二氯甲烷和甲醇的体积比进一步优选为100~80:2,更优选为100:2。
上述配合物的制备方法中,涉及的式(1)、(2)和(3)所示化合物分别为吡啶钌中间体三(2,2'-联吡啶)二氯化钌II、三(1,10-菲咯啉)二氯化钌II和三(4,7-二苯基-1,10-菲咯啉)二氯化钌II,可参考现有文献(Sven Rau,Bernhard AndréGrüβing,Sebastian Schebesta,Katja Lamm,Jana Vieth,Helmar Dirk Walther,ManfredRudolph,Ulrich W.Grummt,Eckard Birkner,Efficient synthesis of rutheniumcomplexes of the type(R-bpy)2RuCl2 and[(R-bpy)2Ru(L–L)]Cl2 by microwave-activated reactions(R:H,Me,tert-But)(L–L:substituted bibenzimidazoles,bipyrimidine,and phenanthroline),Inorganica Chimica Acta 357(2004)4496-4503)进行制备。
上述配合物的制备方法中,涉及的式(L)所示配体可自行设计合成路线进行制备,优选按下述方法进行制备:
取下述式(UAP)所示化合物和二硫化碳置于第二有机溶剂中,在碱性条件下进行缩合反应,制得配体粗品;
在式(L)所示配体的制备方法中,所述式(UAP)所示化合物为熊果酸哌嗪,该化合物可参考现有文献(S.-X.Hua,R.-Z.Huang,M.-Y.Ye,Y.-M.Pan,G.-Y.Yao,Y.Zhang,H.-S.Wang,Design,synthesis and in vitro evaluation of novel ursolic acidderivatives as potential anticancer agents,Eur.J.Med.Chem.95(2015)435-452.)进行合成。相对于式(UAP)所示化合物而言,二硫化碳通常为过量使用。所述的第二有机溶剂优选可以是选自甲醇、乙醇、四氢呋喃、二氧六环、二氯甲烷、氯仿和环己烷中的一种或两种以上的组合。所述第二有机溶剂的用量以能够溶解参加反应的原料为宜,通常情况下,以1mmol的式(UAP)所示化合物为基准,所有参加反应的原料通常用10~20mL的第二有机溶剂来溶解。
在式(L)所示配体的制备方法中,反应体系为pH≥8的碱性条件,为了提高配体的产率,更优选是在pH≥9的条件下进行。可以采用现有常用的碱性物质(如碳酸钠、碳酸钾、碳酸铯、氢氧化钠或氢氧化钾等)来调节体系至碱性,通常是将碱性物质配成水溶液来调节体系至碱性。反应可以在加热或不加热或冰浴条件下进行,因反应较为剧烈,优选是在不加热或冰浴条件下进行。反应过程中采用TLC跟踪监测反应是否完全。根据申请人的经验,当反应在0~25℃条件下进行时,反应时间控制在2~8h较为适宜。
上述式(L)所示配体制备方法制得的是配体粗品,可采用现有常规的纯化方法对其进行纯化以提高配体的纯度。在本申请中,优选采用硅胶柱层析来进行纯化,洗脱时以二氯甲烷和甲醇的组合物为冼脱剂,优选以二氯甲烷和甲醇按200~300:3的体积比组成的组合物为冼脱剂。在洗脱剂的组成中,二氯甲烷和甲醇的体积比进一步优选为230~270:3,更优选为250:3。
本发明还包括上述熊果酸哌嗪二硫代甲酸-吡啶钌配合物或其药学上可接受的盐在制备抗肿瘤药物中的应用。
本发明进一步包括一种药物组合物,含有治疗上有效剂量的上述熊果酸哌嗪二硫代甲酸-吡啶钌配合物或其药学上可接受的盐。
与现有技术相比,本发明提供了三例结构新颖的熊果酸哌嗪二硫代甲酸-吡啶钌配合物及其制备方法和应用,申请人的试验表明,这些配合物对某些肿瘤细胞株具有极为显著的抑制活性,且大大高于其母核和顺铂,同时其对顺铂耐药株细胞(A549-DDP)的抑制活性显著优于常用抗癌药物(如顺铂),有望用于抗肿瘤药物的制备。此外,本发明所述配合物的制备方法简单、反应条件温和,成本低廉。
附图说明
图1为实验例2中配合物I(2μM,24h)和配合物II(2μM,24h)分别与MGC-803细胞相互作用的电镜扫描图,其中(a)表示空白,(b)表示配合物I(2μM)与MGC-803细胞相互作用24h的电镜扫描图(图标尺20μm),(c)为(b)图中标示方框的放大图(图标尺5μm),(d)为(b)图中边缘部分的放大图(图标尺5μm),(e)表示配合物II(2μM)与MGC-803细胞相互作用24h的电镜扫描图(图标尺5μm),(f)表示配合物II(2μM)与MGC-803细胞(与(f)图中的MGC-803细胞不是同一个)相互作用24h的电镜扫描图(图标尺5μm)。
图2为实验例2中配合物I(2μM)和配合物II(2μM)分别对引发MGC-803细胞不同死亡途径的抑制剂的抑制率,其中(a)为配合物I(2μM)分别对引发MGC-803细胞不同死亡途径的抑制剂的抑制率,(b)为配合物II(2μM)分别对引发MGC-803细胞不同死亡途径的抑制剂的抑制率。
图3为实验例2中配合物I(2μM,24h)和配合物II(2μM,24h)分别与MGC-803细胞双染实验荧光显微镜图。
具体实施方式
下面结合具体实施例对本发明作进一步的详述,以更好地理解本发明的内容,但本发明并不限于以下实施例。
在以下实施例或实验例中,对出现的部分简称解释如下:
配合物I或Ru I均表示式(I)所示结构的熊果酸哌嗪二硫代甲酸-吡啶钌配合物。
配合物II或Ru II均表示式(II)所示结构的熊果酸哌嗪二硫代甲酸-吡啶钌配合物。
配合物III或Ru III均表示式(III)所示结构的熊果酸哌嗪二硫代甲酸-吡啶钌配合物。
配体L表示式(L)所示配体。
化合物1表示式(1)所示化合物。
化合物2表示式(2)所示化合物。
化合物3表示式(3)所示化合物。
化合物UAP表示式(UAP)所示化合物。
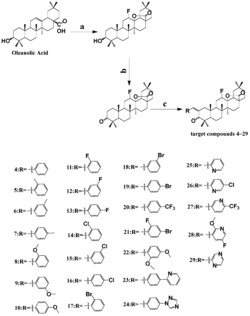
实施例1:配体L的制备
取化合物UAP(5.24g,10mmol,1.0equiv)、4mmol/L氢氧化钠(2.5mL,10mmol,1.0equiv)和二硫化碳(5mL)置于圆底烧瓶中,加入100mL二氯甲烷后(此时体系的pH>9),在室温下搅拌反应4h,停止反应,蒸除溶剂,残余物过硅胶柱层析纯化(二氯甲烷/甲醇=250:3,体积比),得到白色粉末状固体4.53g,产率72.8%。HRMS(m/z)(ESI):C35H55N2NaO2S2[L+Na]
因此,可以确定本实施例所得白色粉末状产物为目标产物配体L(熊果酸哌嗪二硫代甲酸钠),其结构式如下述式(L)所示:
实施例2:配体L的制备
取化合物UAP(2.62g,5mmol,1.0equiv)、4mmol/L氢氧化钠(1.25mL,5mmol,1.0equiv)和二硫化碳(4mL)置于圆底烧瓶中,加入50mL乙醇后(此时体系的pH>9),在室温下搅拌反应2h,停止反应,蒸除溶剂,残余物过硅胶柱层析纯化(二氯甲烷/甲醇=250:3,体积比),得到白色粉末状固体1.82g,产率58.4%。
对本实施例所得产物采用高分辨质谱、核磁共振氢谱、核磁共振碳谱等进行结构表征,确定本实施例所得产物为配体L。
实施例3:配体L的制备
重复实施例2,不同的是,分别用甲醇、四氢呋喃、二氧六环、氯仿或环己烷代替乙醇,采用碳酸钾调节体系的pH=10,最终均得到色粉末状固体。对在不同溶剂中反应所得的产物采用高分辨质谱、核磁共振氢谱、核磁共振碳谱等进行结构表征,确定所得产物均为配体L。
以下实施例按下述合成路线制备配合物I、配合物II和配合物III:
实施例4:配合物I的制备
取配体L(0.622g,1mmol,1.0equiv)、化合物1(0.484g,1mmol,1.0equiv)和四甲基氢氧化铵(25%水溶液)(1mL)置于圆底烧瓶中,加入20mL二氯甲烷-甲醇溶液(v:v=2:1)后,在65下搅拌反应30h,停止反应,蒸除溶剂,残余物经中性氧化铝柱分离纯化(二氯甲烷/甲醇=100:2,体积比),得到白色粉末状固体0.64g,产率63.4%。HRMS(m/z)(ESI):C55H71N6O2RuS2
因此,可以确定本实施例所得白色粉末状产物为目标配合物I,其结构式如下述式(I)所示:
实施例5:配合物I的制备
取配体L(0.311g,0.5mmol,1.0equiv)、化合物1(0.242g,0.5mmol,1.0equiv)和吡啶(0.5mL)置于圆底烧瓶中,加入20mL二氯甲烷-乙醇溶液(v:v=2:1)后,在0℃下搅拌反应24h,停止反应,蒸除溶剂,残余物经中性氧化铝柱分离纯化(二氯甲烷/甲醇=100:2,体积比),得到白色粉末状固体0.271g,产率53.5%。
对本实施例所得产物采用高分辨质谱、核磁共振氢谱、核磁共振碳谱等进行结构表征,确定本实施例所得产物为目标配合物I。
实施例6:配合物II的制备
取配体L(0.622g,1mmol,1.0equiv)、化合物2(0.532g,1mmol,1.0equiv)和四甲基氢氧化铵水溶液(25wt%)(1mL)置于圆底烧瓶中,加入20mL二氯甲烷-甲醇溶液(v:v=2:1)后,在65℃下搅拌反应24h,停止反应,蒸除溶剂,残余物经中性氧化铝柱分离纯化(二氯甲烷/甲醇=100:2,体积比),得到白色粉末状固体0.68g,产率64.1%。HRMS(m/z)(ESI):C59H71N6O2RuS2
因此,可以确定本实施例所得白色粉末状产物为目标配合物II,其结构式如下述式(II)所示:
实施例7:配合物II的制备
取配体L(0.622g,1mmol,1.0equiv)和化合物2(0.532g,1mmol,1.0equiv)置于圆底烧瓶中,加入20mL氯仿-乙二醇甲醚溶液(v:v=1:1)后,在65℃下搅拌反应24h,停止反应,蒸除溶剂,残余物经中性氧化铝柱分离纯化(二氯甲烷/甲醇=100:2,体积比),得到白色粉末状固体0.164g,产率15.5%。
对本实施例所得产物采用高分辨质谱、核磁共振氢谱、核磁共振碳谱等进行结构表征,确定本实施例所得产物为目标配合物II。
实施例8:配合物III的制备
取配体L(0.622g,1mmol,1.0equiv)、化合物3(0.836g,1mmol,1.0equiv)和三乙胺(1mL)置于圆底烧瓶中,加入20mL二氯甲烷-丙醇溶液(v:v=3:1)后,在70℃下搅拌反应24h,停止反应,蒸除溶剂,残余物经中性氧化铝柱分离纯化(二氯甲烷/甲醇=100:2,体积比),得到白色粉末状固体0.892g,产率65.3%。HRMS(m/z)(ESI):C83H87N6O2RuS2[M]
因此,可以确定本实施例所得白色粉末状产物为目标配合物III,其结构式如下述式(III)所示:
实施例9:配合物III的制备
取配体L(0.622g,1mmol,1.0equiv)、化合物3(0.836g,1mmol,1.0equiv)和饱和碳酸钾水溶液(1mL)置于圆底烧瓶中,加入20mL叔丁醇后,在室温下搅拌反应36h,停止反应,蒸除溶剂,残余物经中性氧化铝柱分离纯化(二氯甲烷/甲醇=100:2,体积比),得到白色粉末状固体0.417g,产率30.6%。
对本实施例所得产物采用高分辨质谱、核磁共振氢谱、核磁共振碳谱等进行结构表征,确定本实施例所得产物为目标配合物III。
实验例1:本发明目标配合物对多种人肿瘤细胞株的体外抗肿瘤活性实验:
为说明本发明所述熊果酸哌嗪二硫代甲酸-吡啶钌配合物的抗肿瘤作用,申请人对三个配合物均进行了抗肿瘤活性实验(以常用金属基抗肿瘤药物顺铂(Cis-platin)为参比)。
采用MTT法测试化合物的体外抗肿瘤活性。取处于对数生长期的细胞,每孔180μL(约4500-5000个细胞)含细胞的培养基接种于96孔培养板,于37℃、5%CO2充分湿化条件下培养24h。待细胞贴壁后,按每孔20μL的量加入样品,每个样品设6个复孔,同时设定相应的空白对照。继续培养48h后,每孔加入10μL MTT试剂(浓度为5mg/mL),继续孵育4h后,吸弃上清液,每孔再加入150μL DMSO,轻微震荡反应5~8min,使结晶颗粒充分溶解。空白对照组调零,用酶标仪以490nm波长测定去除本底光吸收值后的吸光度值( 值),计算细胞增殖抑制率,对初筛抗肿瘤效果好的受试化合物,继续用5个浓度梯度继续做相应细胞株的IC50值,所有实验均重复3次后取平均值。实验结果详见下表1和表2。
由表1中数据可知,本发明所述配合物I和II对人胃癌细胞MGC-803、人膀胱癌细胞T24、人肝癌细胞HePG2、人鼻咽癌细胞CNE2、人乳腺癌细胞MDA-MB-231和MCF-7、人肺腺癌细胞株A549等抑制活性显著优于常用其母核及抗肿瘤药物顺铂;本发明所述配合物III对人膀胱癌细胞T24、人乳腺癌细胞MDA-MB-231和MCF7等抑制活性显著优于常用抗肿瘤药物顺铂。以上结果说明,通过将熊果酸活性配体引入到吡啶基钌结构上制备出新型的熊果酸哌嗪二硫代甲酸-吡啶钌抗肿瘤配合物是可行的,可通过金属与配体的协同效应筛选出高效的新型抗肿瘤配合物。
表1:化合物对不同肿瘤细胞株的半抑制率浓度(IC50,μM).
表2:化合物对A549细胞株和A549-DDP耐药细胞株的半抑制率浓度(IC50,μM).
由表2中数据可知,顺铂对人肺腺癌耐顺铂株A549-DDP的抑制活性与人肺腺癌非顺铂耐药株A549相比,显著减小了,说明顺铂具有明显的耐药性;而配合物I、II和III对顺铂耐药株细胞的A549-DDP抑制活性与非顺铂耐药株A549相比,没有太大的变化,说明配合物I、II和III对顺铂耐药株A549-DDP没有明显的耐药性。
以上结果说明,通过将熊果酸活性配体引入到吡啶基钌结构上制备出新型的熊果酸哌嗪二硫代甲酸-吡啶钌抗肿瘤配合物可望改善抗肿瘤配合物的耐药性。
实验例2:本发明所述配合物的抗肿瘤作用机制
为说明本发明所述熊果酸哌嗪二硫代甲酸-吡啶钌配合物的抗肿瘤作用机制,申请人先通过电镜扫描配合物I和II与肿瘤细胞MGC-803的相互作用情况,结果如图1所示。
由图1可知,与空白相比,配合物I和II的加入可以引发囊泡(标示的小方框)和内凹(标示的箭头),说明这两个配合物可以通过内吞作用机制发挥抗肿瘤作用。
为了进一步阐明配合物引发细胞死亡的模式,申请人将细胞坏死抑制剂Necrostatin-1、与溶酶体相关的蛋白Leupeptin、细胞凋亡抑制剂Z-VAD-FMK、类凋亡抑制剂Cycloheximide和细胞自噬抑制剂3-Methyladenine(3-MA)与MGC-803细胞株、配合物I和II相互作用,结果如图2所示。
由图2可知,配合物I和II主要是通过坏死以及与溶酶体相关的通路发挥抗肿瘤作用。
通过Hochest 33258-PI双重染色实验的结果(如图3所示),与不加药对照组比较,配合物I和II在诱导MGC-803细胞发生死亡时,细胞主要发生晚凋或者坏死,也反证了图2的实验结果。
熊果酸哌嗪二硫代甲酸-吡啶钌配合物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0