








专利摘要
本发明提供一种ZrOxNy陶瓷先驱体的制备方法,该方法以金属锆化合物为锆源,以胺类有机物为氮源,以氧类有机物为氧源和溶剂,以三乙胺为催化剂,通过共聚反应制备ZrOxNy陶瓷先驱体。利用本发明制得的ZrOxNy先驱体具有较高的陶瓷收率。
权利要求
1.一种ZrO
将金属锆化合物溶解后与胺类有机物混合后,发生共聚反应得到所述ZrO
所述金属锆化合物为 ZrCl
所述金属锆化合物、所述胺类有机物、催化剂与溶剂按摩尔比为1:0.1~10:2~3:10~2000混合进行所述共聚反应;
所述催化剂为三乙胺;
所述溶剂为四氢呋喃、乙醇、二甲亚砜或N,N-二甲基甲酰胺中任一种。
2.根据权利要求1所述的ZrO
3.根据权利要求1所述的ZrO
4.根据权利要求1~3中任一项所述的ZrOxNy陶瓷先驱体的制备方法,其特征在于,所述金属锆化合物、所述胺类有机物、所述催化剂与所述溶剂按摩尔比为1:0.5~2:2~3:50~200混合进行所述共聚反应。
5.根据权利要求4所述的ZrO
6.一种如权利要求1~5中任一项所述的ZrO
7.根据权利要求6所述制备ZrO
8.根据权利要求7所述制备ZrO
说明书
技术领域
本发明涉及耐高温陶瓷技术领域,具体的涉及一种ZrOxNy陶瓷先驱体的制备方法。
背景技术
近年来,过渡金属氮氧结构MOxNy陶瓷引起了广泛的关注,特别是ZrOxNy陶瓷在催化剂领域的研究。实验研究表明,ZrOxNy陶瓷可用于催化氨气的分解制备氢气和氮气。当温度为 550℃时,ZrOxNy陶瓷具有催化活性,β-Zr7O11N2陶瓷转变为富氮β-ZrOxNy(Zr7O9,5N3)陶瓷,进而催化氨气的分解。此外,ZrOxNy陶瓷的使用避免了热解氨气过程中联肼的生成。结果表明,ZrOxNy陶瓷具有非均相催化性能(H.Soerijanto,C. U.Wildb,M.Lerch,R. R. T.Ressler.The impact of nitrogen mobility on the activityof zirconium oxynitride catalysts for ammonia decomposition.Journal ofCatalysis.2007,250:19–24)。热解制备的N掺杂ZrO2具有较高纯度的单斜晶型。N掺杂ZrO2催化剂的光学特性明显改变,N掺杂后,在可见光和近红外区域均有光学响应,同时在UVA区域出现一个强吸收峰。与ZrO2相比,在降解染料(AM和MB)方面,N掺杂ZrO2具有更高的催化活性(H.Sudrajat,S.Babel,H.Sakai,S. Takizawa.Rapid enhanced photocatalyticdegradation of dyes using novelN-doped ZrO2.Journal of EnvironmentalManagement.2016,165:224-234.)。因此,ZrOxNy陶瓷以其优异的催化性能受到了研究人员的关注,在非均相催化领域具有广阔的应用前景。此外,过渡金属IVB氧氮化物也具有优异的耐腐蚀性能(包括酸性和氧化环境),被用于陶瓷薄膜、涂层和电池电极等领域。
通常采用气氛烧结法和磁控溅射法制备ZrOxNy陶瓷。其中,气氛烧结法是将ZrO2陶瓷在氨气或氮气气氛烧成或ZrN在氧气等含氧气氛中烧成制备ZrOxNy陶瓷的方法,但该制备方法难以得到纯度较高的ZrOxNy陶瓷。磁控溅射法是以单质金属、金属氯化物ZrClx或金属氧化物作为金属源,N2/O2、N2/H2O或Air作为氮源和氧源,通过溅射制备ZrOxNy陶瓷的方法,但该技术存在对设备要求高、能耗高、制备工艺复杂以及制备的陶瓷组份不纯等缺点。同时采用现有的制备方法还存在难以通过简单的工艺制备具有单一组份的ZrOxNy陶瓷。
发明内容
本发明的目的在于提供一种ZrOxNy陶瓷先驱体的制备方法,该发明解决了单一组份 ZrOxNy陶瓷制备设备投入大、制备过程复杂,采用简单方法难以制得单一组份陶瓷的技术问题。
本发明的一方面还提供了提供一种ZrOxNy陶瓷先驱体的制备方法,包括以下步骤:将有机金属锆化合物溶解后与胺类有机物混合后,发生共聚反应得到ZrOxNy陶瓷先驱体,其中 x=1.0000~1.5714,y=0.2857~1.0000;有机金属锆化合物、胺类有机物、催化剂与溶剂按摩尔比为1:0.1~10:2~3:10~2000混合进行共聚反应;催化剂为三乙胺;溶剂为四氢呋喃、乙醇、二甲亚砜或N,N-二甲基甲酰胺中任一种。
进一步地,共聚反应条件为在-30~150℃下,反应0.5~10小时。
进一步地,还包括对ZrOxNy陶瓷先驱体进行蒸馏除去溶剂,蒸馏在30-250℃下进行。
进一步地,有机金属锆化合物、胺类有机物、催化剂与溶剂按摩尔比为1:0.5~2:2~3:50~200 混合进行共聚反应。
进一步地,有机金属锆化合物为ZrCl4、ZrOCl2·8H2O、Cp2ZrCl2或(R2N)2ZrCl2中任一种,其中R=甲基、乙基或丙基。
进一步地,胺类有机物为尿素、单氰胺、双氰胺或三聚氰胺中任一种。
本发明另一方面还提供了一种如上述的ZrOxNy陶瓷先驱体用于制备ZrOxNy陶瓷的应用。
进一步地,ZrOxNy陶瓷的制备条件为:加热ZrOxNy陶瓷先驱体至500~1600℃,裂解0.5~5 小时。
进一步地,加热步骤在通氨气环境中进行。
本发明的技术效果:
1、本发明提供ZrOxNy陶瓷先驱体的制备方法原料易得、价格低廉、成本低;用于制备 ZrOxNy陶瓷时制备过程简单。
2、将本发明所制备的先驱体通过烧结处理即可得到组份单一的且纯度较高的ZrOxNy陶瓷,即可以用于制备Zr2ON2、Zr7O8N4和Zr7O11N2等陶瓷。
具体请参考根据本发明的ZrOxNy陶瓷先驱体的制备方法提出的各种实施例的如下描述,将使得本发明的上述和其他方面显而易见。
附图说明
图1为本发明优选实施例1中所得先驱体的红外光谱图;
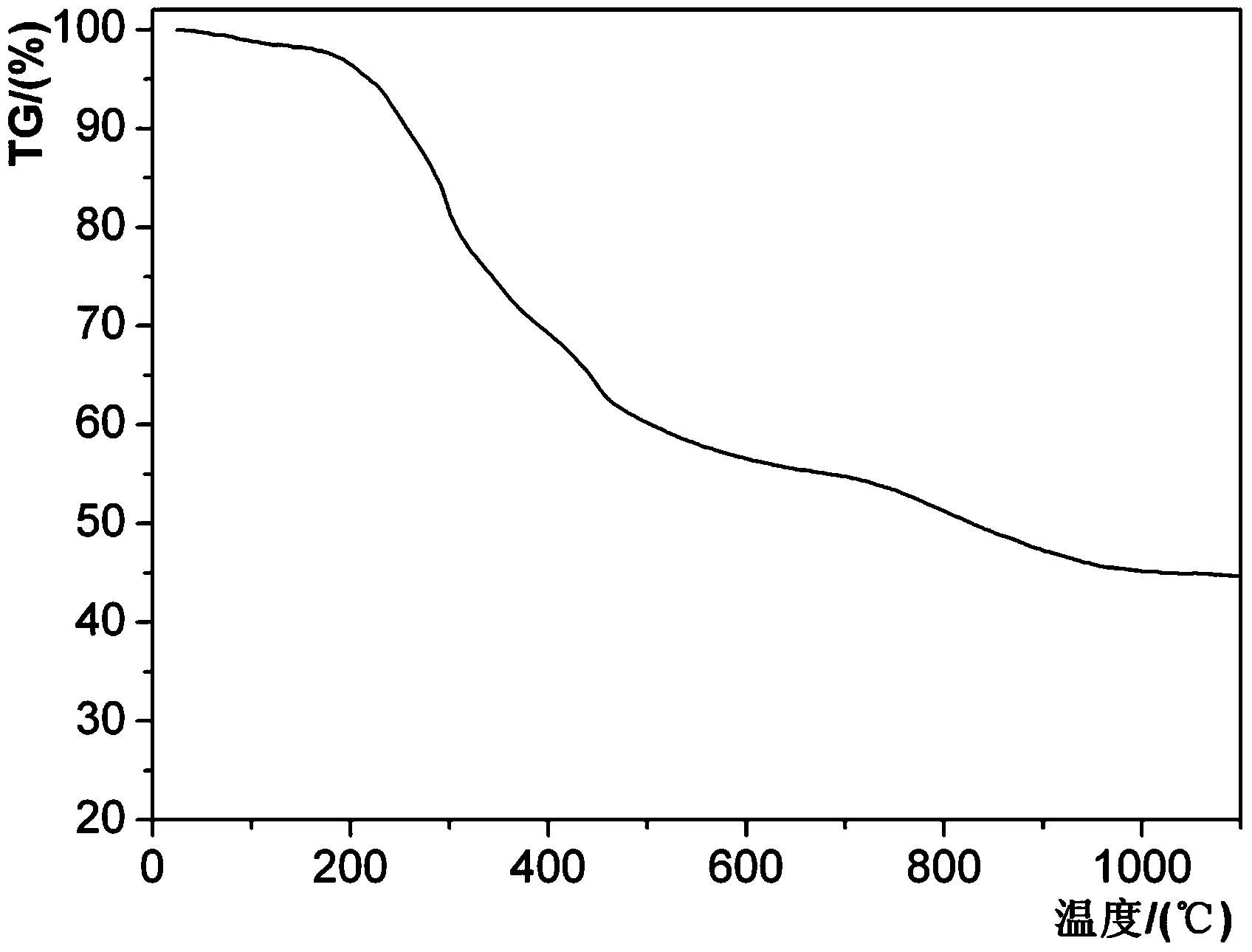
图2为本发明优选实施例1中所得先驱体的热失重曲线图;
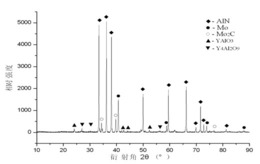
图3为本发明优选实施例1中所得先驱体制备得到的Zr2ON2陶瓷的XRD谱图。
具体实施方式
构成本申请的一部分的附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
本发明提供的ZrOxNy陶瓷先驱体的制备方法,包括以下步骤:
将有机金属锆化合物溶解后与胺类有机物混合后,发生共聚反应得到ZrOxNy陶瓷先驱体,其中x=1.0000~1.5714;y=0.2857~1.0000;有机金属锆化合物、胺类有机物、催化剂与溶剂按摩尔比为1:0.1~10:2~3:10~2000混合进行共聚反应;催化剂为三乙胺;溶剂为四氢呋喃、乙醇、二甲亚砜或N,N-二甲基甲酰胺中任一种。在溶解状态下的有机金属锆化合物中加入胺类有机物,从而使得所制得的ZrOxNy陶瓷先驱体用于制备ZrOxNy陶瓷时,能有效提高所得ZrOxNy陶瓷的陶瓷产率至45%。该反应以含有氧的溶剂作为氧源,通过有机金属锆化合物与胺类有机物在催化剂存在条件下进行共聚反应,产物产量较高。
按此比例混合进行有机金属锆化合物、胺类有机物与溶剂,能提高反应程度,提高所得产物中所制备ZrOxNy陶瓷中的比例,减少杂质含量。制得具有单一组份的ZrOxNy陶瓷。从对所得先驱体进行的红外光谱图和XRD图中可以看出。更优选的,有机金属锆化合物、胺类有机物、催化剂与溶剂按摩尔比为1:0.5~2:2~3:50~200混合进行共聚反应。按此比例混合,所得共聚产物收率高和裂解产物ZrOxNy的比例达到最高。
本发明中所用有机金属锆化合物与胺类有机物可为各类常用于制备ZrOxNy陶瓷的有机金属锆化合物与胺类有机物。例如可以为有机金属锆化合物为ZrCl4、ZrOCl2·8H2O、Cp2ZrCl2或(R2N)2ZrCl2,其中R=甲基、乙基或丙基。胺类有机物为尿素、单氰胺、双氰胺或三聚氰胺。
有机金属锆化合物的溶解条件为在惰性气氛保护下,-60~60℃下溶解。按此条件能保证有机金属锆化合物充分溶解。
优选的,共聚反应条件为在-30~150℃下,反应0.5~10小时。按此条件进行反应,共聚产物收率较高,可达95%。能提高所得产物中ZrOxNy的比例,减少杂质的产生。
优选的,还包括对ZrOxNy陶瓷先驱体进行蒸馏除去溶剂,蒸馏在30-250℃下进行。经过蒸馏后能回收所得溶剂,降低生产成本。
优选的,有机金属锆化合物为ZrCl4、ZrOCl2·8H2O、Cp2ZrCl2或(R2N)2ZrCl2中任一种,其中R=甲基、乙基或丙基。优选的,胺类有机物为尿素、单氰胺、双氰胺或三聚氰胺中任一种
本发明的另一方面还提供了上述ZrOxNy陶瓷先驱体用于制备ZrOxNy陶瓷的应用。
优选的,ZrOxNy陶瓷的制备条件为:加热ZrOxNy陶瓷先驱体至500~1600℃,裂解0.5~5 小时。以先驱体为裂解对象,能提高所得产物的陶瓷产率。裂解可以在各类气氛中进行,例如惰性气氛或氨气环境中进行。更优选的,裂解温度为700~1300℃。
优选的,加热步骤在通氨气环境中进行。通以氨气,能将加热裂解过程中产生的自由碳除去,提高裂解后所得产物组份的单一性。
实施例
以下实施例中所用物料或仪器均为市售。
实施例1
1、将0.05molZrCl4、0.15mol三乙胺和1.00mol N,N-二甲基甲酰胺混合后,将温度控制在 20℃左右并搅拌直至溶解完毕,继续30分钟;
将0.06mol双聚氰胺缓慢加到上述溶液中,加热至80℃继续搅拌10h;
将反应器温度调至250℃,减压蒸馏除去溶剂和三乙胺盐酸盐,冷却至室温,得到黄色的陶瓷先驱体;
2、将陶瓷先驱体装入氧化铝坩埚,放入高温裂解炉,在氩气气氛下以5℃/分钟升温至 500~1600℃后,保温裂解2小时,自然冷却后可分别得到Zr2ON2、Zr7O8N4和Zr7O11N2等ZrOxNy结构陶瓷产物。
图1是本发明实施例制备所得先驱体的红外光谱图,图2是本发明实施例制备所得先驱体的热失重曲线图,图3本发明实施例制备所得先驱体制备Zr2ON2陶瓷的XRD谱图。
由图1可知,先驱体结构中含有Zr-N、C=N和CΞN等化学键。
由图2可知,将所得先驱体在900℃热处理后的Zr2ON2陶瓷的产率可达45%。
由图3可知,制备的Zr2ON2陶瓷纯度较高,无其他杂质峰,其中以ZrOxNy含量为主,实现了组份单一。
实施例2
1、将0.05molCp2ZrCl2、0.15mol三乙胺和1.00mol N,N-二甲基甲酰胺混合后,将温度控制在20℃左右并搅拌直至溶解完毕,继续30分钟;
将0.06mol尿素缓慢加到上述溶液中,加热至80℃继续搅拌10h;
将反应器温度调至250℃,减压蒸馏除去溶剂和三乙胺盐酸盐,冷却至室温,得到陶瓷先驱体;
2、将陶瓷先驱体装入氧化铝坩埚,放入高温裂解炉,在氩气气氛下以5℃/分钟升温至 900℃后,保温裂解2小时,自然冷却后得到Zr2ON2陶瓷产物。陶瓷产率为35%。
实施例3
1、将0.05mol(Et2N)2ZrCl2、0.15mol三乙胺和1.00mol N,N-二甲基甲酰胺混合后,将温度控制在20℃左右并搅拌直至溶解完毕,继续30分钟;
将0.05mol三聚氰胺缓慢加到上述溶液中,加热至80℃继续搅拌10h;
将反应器温度调至250℃,减压蒸馏除去溶剂和三乙胺盐酸盐,冷却至室温,得到黄色的陶瓷先驱体;
2、将陶瓷先驱体装入氧化铝坩埚,放入高温裂解炉,在氩气气氛下以5℃/分钟升温至 900℃后,保温裂解2小时,自然冷却后得到Zr2ON2陶瓷产物。陶瓷产率为38%。
实施例4
与实施例1的区别仅在于:有机金属锆化合物(ZrOCl2·8H2O)、胺类有机物、催化剂与溶剂按摩尔比为1:0.1:2:10;有机金属锆化合物的溶解条件为在惰性气氛保护下,-60℃下进行溶解;共聚反应条件为在-30℃下,反应0.5小时;ZrOxNy陶瓷先驱体进行蒸馏除去溶剂和三乙胺盐酸盐,蒸馏在250℃下进行;ZrOxNy陶瓷的制备条件为:在惰性气氛下加热ZrOxNy陶瓷先驱体至500℃,裂解0.5小时。所得先驱体的裂解条件为1300℃。
实施例5
与实施例1的区别仅在于:有机金属锆化合物((R2N)2ZrCl2,R=甲基)、胺类有机物、催化剂与溶剂按摩尔比为1:10:3:2000;有机金属锆化合物的溶解条件为在惰性气氛保护下,60℃下进行溶解;共聚反应条件为在150℃下,反应10小时;ZrOxNy陶瓷先驱体进行蒸馏除去溶剂,蒸馏在160℃下进行;ZrOxNy陶瓷的制备条件为:在惰性气氛下加热ZrOxNy陶瓷先驱体至1000℃,裂解5小时。溶剂为二甲亚砜。所得先驱体的裂解条件为500℃下0.5小时。
实施例6
与实施例1的区别仅在于:有机金属锆化合物((R2N)2ZrCl2,R=乙基)、胺类有机物、催化剂与溶剂按摩尔比为1:2:3:200。胺类有机物为单氰胺。所得先驱体的裂解条件为1600℃下 5小时。
实施例7
与实施例1的区别仅在于:有机金属锆化合物((R2N)2ZrCl2,R=丙基)、胺类有机物、催化剂与溶剂按摩尔比为1:0.52:50。胺类有机物为双氰胺。所得先驱体的裂解条件为700℃。
本领域技术人员将清楚本发明的范围不限制于以上讨论的示例,有可能对其进行若干改变和修改,而不脱离所附权利要求书限定的本发明的范围。尽管己经在附图和说明书中详细图示和描述了本发明,但这样的说明和描述仅是说明或示意性的,而非限制性的。本发明并不限于所公开的实施例。
通过对附图,说明书和权利要求书的研究,在实施本发明时本领域技术人员可以理解和实现所公开的实施例的变形。在权利要求书中,术语“包括”不排除其他步骤或元素,而不定冠词“一个”或“一种”不排除多个。在彼此不同的从属权利要求中引用的某些措施的事实不意味着这些措施的组合不能被有利地使用。权利要求书中的任何参考标记不构成对本发明的范围的限制。
一种ZrOxNy陶瓷先驱体的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0