






专利摘要
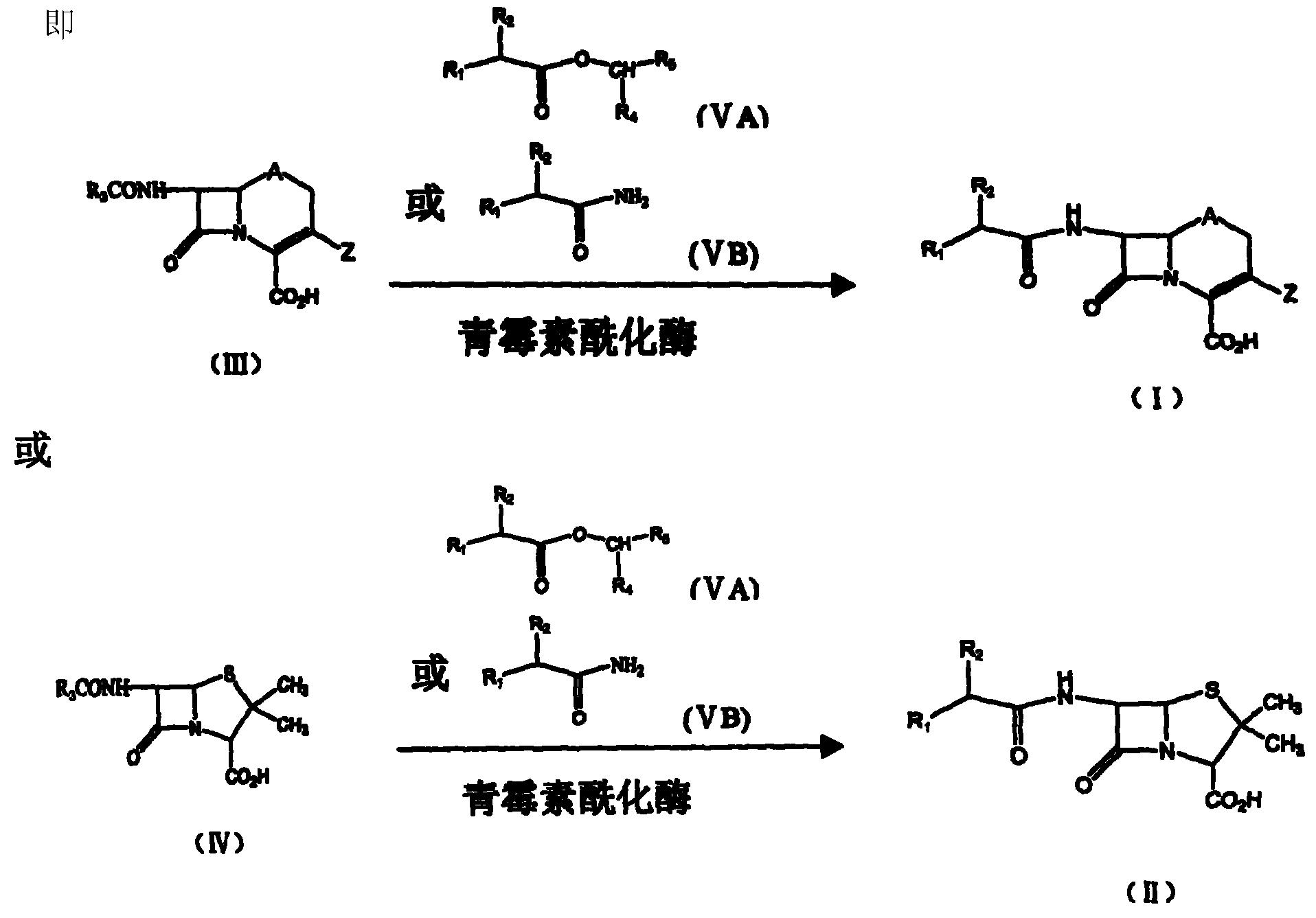
本发明涉及抗生素的制备方法,更具体地说,涉及采用酶催化“一锅法”制备β-内酰胺抗生素(即头孢菌素和青霉素)的方法。在水或水与醇混合物介质中,在游离或/和固定化的青霉素酰化酶存在下,通式(III)化合物与通式(VA)的酯或通式(VB)的酰胺反应,得到通式(I)的头孢菌素;或通式(IV)化合物与通式(VA)的酯或通式(VB)的酰胺反应,得到通式(II)的青霉素。
权利要求
1.一种酶催化“一锅法”制备β-内酰胺抗生素的方法,其特征是,该方法包括:在水或水与醇混合物反应介质中,在游离或/和固定化的青霉素酰化酶存在下,控制pH为5.0~9.0,通式(III)化合物与通式(VA)的酯或通式(VB)的酰胺反应,得到通式(I)的头孢菌素;或通式(IV)化合物与通式(VA)的酯或通式(VB)的酰胺反应,得到通式(II)的青霉素;
或
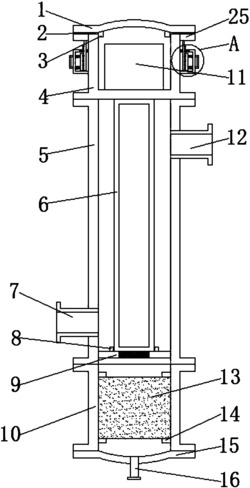
其中,A表示硫原子或氧原子;
Z表示氢原子、卤素原子、羟基、低级烷氧基、取代或非取代低级烷基、C2-C4链烯基、杂环硫亚甲基或杂环亚甲基;
R1为取代或非取代的6元环基或杂环基;
R2表示氨基或氢原子;
R3表示芳基亚甲基或芳氧基亚甲基;
R4为氢、CH2X或CHXY,R5为氢、CH2Y或CHXY,其中X和Y各自独立地代表氢、羟基、低级烷基或羟基-低级烷基。
2.根据权利要求1所述的制备β-内酰胺抗生素的方法,其特征是,所述的R1为苯基、对羟基苯基、双氢苯基或1H-四氮唑基。
3.根据权利要求1所述的制备β-内酰胺抗生素的方法,其特征是,所述的通式(III)或(IV)化合物与通式(VA)的酯或通式(VB)的酰胺的摩尔比为1∶1~10。
4.根据权利要求3所述的制备β-内酰胺抗生素的方法,其特征是,所述的通式(III)或(IV)化合物与通式(VA)的酯或通式(VB)的酰胺的摩尔比为1∶1.5~3。
5.根据权利要求1所述的制备β-内酰胺抗生素的方法,其特征是,所述的水或水与醇混合物介质与通式(III)或(IV)化合物,按重量比为5~200∶1。
6.根据权利要求1所述的制备β-内酰胺抗生素的方法,其特征是,所述的水与醇混合物,其中所述的醇为甲醇、乙醇、乙二醇或丙三醇,醇在水与醇混合物中含量为0~50wt.%。
7.根据权利要求1所述的制备β-内酰胺抗生素的方法,其特征是,所述的青霉素酰化酶的用量为通式(III)或(IV)化合物的0.1~100倍,以重量计。
8.根据权利要求1所述的制备β-内酰胺抗生素的方法,其特征是,所述的青霉素酰化酶是固定化的青霉素酰化酶。
9.根据权利要求1所述的制备β-内酰胺抗生素的方法,其特征是,该方法在-5℃~40℃的温度下进行。
10.根据权利要求1所述的制备β-内酰胺抗生素的方法,其特征是,所述的pH为6.5~7.5。
说明书
技术领域技术领域
本发明涉及抗生素的制备方法,更具体地说,涉及采用酶催化“一锅法”制备β-内酰胺抗生素(即头孢菌素和青霉素)的方法。
技术背景背景技术
β-内酰胺抗生素,即头孢菌素和青霉素,是临床上广泛使用的抗生素。头孢菌素的结构式如通式(I)所示,青霉素的结构式如通式(II)所示:
其中,A表示硫原子或氧原子;
Z表示氢原子、卤素原子、羟基、低级烷氧基、取代或非取代低级烷基、C2-C4链烯基、杂环硫亚甲基或杂环亚甲基;
R1为取代或非取代的6元环基或杂环基;例如苯基、对羟基苯基、双氢苯基或1H-四氮唑基;
R2表示氨基或氢原子。
在临床上应用的头孢克洛,即通式(I)中,Z为氯原子,R1为苯基,R2为氨基;在临床上应用的头孢丙烯或E-型头孢丙烯,即通式(I)中,Z为丙烯基,R1为对羟基苯基,R2为氨基;在临床上应用的头孢氨苄,即通式(I)中,Z为甲基,R1为苯基,R2为氨基;在临床上应用的头孢拉定,即通式(I)中,Z为甲基,R1为双氢苯基,R2为氨基;在临床上应用的头孢羟氨苄,即通式(I)中,Z为甲基,R1为对羟基苯基,R2为氨基;在临床上应用的头孢唑啉,即通式(I)中,Z为(2-甲基-1,3,4-噻二唑-5-基)硫亚甲基,R1为1H-四氮唑基,R2为氢原子。
在临床上应用的氨苄西林,即通式(II)中,R1为苯基,R2为氨基;在临床上应用的阿莫西林,即通式(II)中,R1为对羟基苯基,R2为氨基。
已有很多文献报道采用酶法来制备β-内酰胺抗生素。采用酶法制备β-内酰胺抗生素的优点是,避免使用有机溶剂,环境污染小,而且反应条件比较温和,操作简便,成本低。
WO9920786公开了一种酶法合成β-内酰胺抗生素的方法,6-氨基青霉烷酸(即下面通式(VII)化合物)或7-氨基头孢烷酸(即下面通式(VI)化合物)与酰化试剂进行酶酰化反应,得到相应的青霉素或头孢菌素。在该方法中,其特征在于,6-氨基青霉烷酸或7-氨基头孢烷酸和酰化试剂处于过饱和浓度。
WO9804732公开了一种合成β-内酰胺抗生素的方法。该方法是在酰化酶存在下,6-氨基青霉烷酸或7-氨基头孢烷酸与酯反应,得到相应的青霉素或头孢菌素。在该方法中,采用酯作为酰化试剂。
在上述方法中,作为起始原料的7-氨基头孢烷酸(即通式(VI)化合物),可以由下述通式(III)化合物通过7位处侧链的酰氨基水解脱去R3CO-,分离后得到;6-氨基青霉烷酸(即通式(VII)化合物),可以由下述通式(IV)化合物通过6位处侧链的酰氨基水解脱去R3CO-,分离后得到。在分离得到7-氨基头孢烷酸或6-氨基青霉烷酸后,按照WO9920786或WO9804732所述的方法制备各种头孢菌素或青霉素。如果能将这两步反应合并在同一反应体系中,连续进行,即在“一锅”中,在水解脱去R3CO-的同时酶酰化结合上头孢菌素或青霉素所需的活性基团R1R2CO-,这样简化了反应步骤,避免了7-氨基头孢烷酸或6-氨基青霉烷酸中间产物的分离,大大缩短了反应流程,降低成本。
发明内容发明内容
因此,本发明的目的在于,提供一种在同一反应体系中、在酶作用下水解和酰化缩合同时进行的酶催化“一锅法”制备β-内酰胺抗生素的方法。
本发明提供的酶催化“一锅法”制备β-内酰胺抗生素的方法,该方法包括:在水或水与醇混合物反应介质中,在游离或/和固定化的青霉素酰化酶存在下,通式(III)化合物与通式(VA)的酯或通式(VB)的酰胺反应,得到通式(I)的头孢菌素;或通式(IV)化合物与通式(VA)的酯或通式(VB)的酰胺反应,得到通式(II)的青霉素;
或
其中,A表示硫原子或氧原子;
Z表示氢原子、卤素原子、羟基、低级烷氧基、取代或非取代低级烷基、C2-C4链烯基、杂环硫亚甲基或杂环亚甲基;
R1为取代或非取代的6元环基或杂环基;例如苯基、对羟基苯基、双氢苯基
或1H-四氮唑基;
R2表示氨基或氢原子;
R3表示芳基亚甲基或芳氧基亚甲基;
R4为氢、CH2X或CHXY,R5为氢、CH2Y或CHXY,其中X和Y各自独立地代表氢、羟基、低级烷基或羟基-低级烷基。
本说明书中所示各基团分别具体如下所述。卤素原子是指氟、氯、溴或碘。低级烷氧基,例如甲氧基、乙氧基、丙氧基等。取代或非取代低级烷基,低级烷基是指例如甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基等直链或支链状的C1~C4烷基;卤代的低级烷基,例如氯甲基、溴甲基、碘甲基等;烷氧基取代的低级烷基,例如甲氧基甲基、乙氧基甲基等;以及乙酰氧基甲基、氨基甲酰氧基甲基等。C2-C4链烯基,例如乙烯基、丙烯基、2,2-二溴乙烯基等。杂环硫亚甲基例如1,2,3-三唑-4-基硫亚甲基、5-甲基-1,3,4-噻二唑-2-基硫亚甲基、3-甲基-1,3,4-三嗪-5,6-二酮-2-硫亚甲基、1-甲基四唑-5-基硫亚甲基、1-(2-二甲氨基乙基)四唑-5-基硫亚甲基、1,3,4-噻二唑-5-基硫亚甲基、1-(2-羟基乙基)四唑-5-基硫亚甲基等;杂环亚甲基例如1-甲基吡咯烷基亚甲基、吡啶亚甲基、1,2,3-三唑基、1,5-环戊二烯基-吡啶亚甲基等。
R3所述的芳基亚甲基或芳氧基亚甲基,其中,芳基是指苯基、茴香基、萘基等。R3,例如为苄基、甲苯基亚甲基、二甲苯基亚甲基、萘基亚甲基或对甲氧基苄基等。
在本发明所提供的制备方法中,在R2为氢原子时,R1优选为6元杂环基,例如1H-四氮唑基。
在本发明中,用作起始原料的通式(III)所示的β-内酰胺化合物,为已知化合物,可以按照Torii等在四面体快报,Tetrahedron Lett,23,2187~2188(1982)叙述的方法来制备3-卤代头孢烯化合物;在制备了3-卤代头孢烯化合物之后,可按照CN1257508A或中国专利申请03146092.5所述的方法,通过亲核取代反应或Wittig等反应,在3位C上引入所需要的取代基;然后,按照J.O.C.1991,56,3638所述方法在酚类化合物作用下,断4位上的羧基保护基,来制得通式(III)所示的β-内酰胺化合物。
用作起始原料的通式(IV)所示的β-内酰胺化合物,可市购得到。
通式(VA)的酯或通式(VB)的酰胺,可市购得到,或者按WO9804732所述方法来制备。
所述的青霉素酰化酶,可以是游离的或固定化的青霉素酰化酶,优选固定化的青霉素酰化酶,例如可购买到的德国Bringer公司PGA-450,中国长沙福来格公司IPA-750等,酶菌来源,例如Acetobacter pasterurianum、Alcaligenes faecalis、Bacillus megaterium、Escherichia coli、Fusariumoxysporum或Xanthomonas citrii。
在本发明所提供的方法中,在-5℃~40℃的温度下,在水或水与醇混合物介质中,通过滴加碱使通式(III)或(IV)化合物溶解,加入通式(VA)的酯或通式(VB)的酰胺,采用酸或碱使该反应体系的pH为5.0~9.0,再向该体系中加入游离或固定化的青霉素酰化酶。在反应过程中,用酸或碱维持该反应体系的pH为5.0~9.0,优选6.0~8.0,更优选6.5~7.5。
本发明方法中,采用水或水与醇混合物为反应介质,其中醇可以为一元醇、二元醇或三元醇。一元醇,例如甲醇或乙醇;二元醇,例如乙二醇;三元醇,例如丙三醇。醇的用量为,醇在水与醇混合物中含量为0~50wt.%。
在本发明中,用来调节体系pH的酸可以为无机酸或有机酸,其中所述的无机酸包括盐酸、磷酸或硫酸,优选盐酸;所述的有机酸包括甲酸或乙酸。用来调节体系pH的碱包括无机碱或有机碱,其中所述的无机碱包括氢氧化钠溶液、氢氧化钾溶液或氨水等;所述的有机碱包括三乙胺或正丁胺等。在本发明方法中,优选氢氧化钠溶液、氨水或三乙胺。
在该方法中,通式(III)或(IV)化合物与通式(VA)的酯或通式(VB)的酰胺的摩尔比为1∶1~10,优选1∶1.5~3;水或水与醇的混合物反应介质与通式(III)或(IV)化合物,按重量比为5~200∶1的量,更优选10~20∶1;所述的青霉素酰化酶的用量通常根据酶的活性而定,一般为通式(III)或(IV)化合物的0.1~100倍,以重量计。
在本发明所提供的方法中,通常在-5℃~40℃的温度下进行,优选5℃~20℃;整个反应时间通常为2~18小时。
在反应完成后,可以采用常规已知方法分离通式(I)的头孢菌素或通式(II)的青霉素,例如采用与β-萘酚形成复合物的方法。
在水或水与醇混合物介质中,在青霉素酰化酶存在下,通式(III)化合物在7位酰胺基水解脱去R3CO-,生成通式(VI)的7-氨基头孢烷酸(高效液相色谱检测出),而同时通式(VA)的酯或通式(VB)的酰胺在酶的作用下与生成的7-氨基头孢烷酸缩合,生成通式(I)的头孢菌素。而通式(IV)化合物在6位酰胺基水解脱去R3CO-,生成通式(VII)的6-氨基青霉烷酸(高效液相色谱检测出),而同时通式(VA)的酯或通式(VB)的酰胺在酶的作用下与生成的6-氨基青霉烷酸缩合,生成通式(II)的青霉素。
因此,本发明提供的酶催化“一锅法”制备β-内酰胺抗生素的方法,在同一反应体系中水解和缩合同时进行,避免了7-氨基头孢烷酸或6-氨基青霉烷酸中间产物的分离,这样简化了反应步骤,大大缩短了反应流程,降低成本。而且反应体系不形成粘稠物,产品溶液与固定化酰胺化酶易于分离。
附图说明具体实施方式具体实施方式
下面提供的实施例是用来具体地说明本发明的实施方案,这些实施例不限制本发明的范围。
在本发明中,根据《美国药典》(25版,第351页至352页)规定的测定方法,采用高效液相色谱法测定头孢丙烯的生成量;根据《中国药典》(2000版,第175页至176页)规定的测定方法,采用高效液相色谱法测定头孢克洛的生成量;根据《中国药典》(2000版,第190页至191页)规定的测定方法,采用高效液相色谱法测定头孢氨苄的生成量;根据《中国药典》(2000版,第182页至183页)规定的测定方法,采用高效液相色谱法测定头孢拉啶的生成量;根据《中国药典》(2000版,第193页至194页)规定的测定方法,采用高效液相色谱法测定头孢羟氨苄生成量;根据《中国药典》(2000版,第189页)规定的测定方法,采用高效液相色谱法测定头孢唑啉生成量;根据《中国药典》(2000版,第717页)规定的测定方法,采用高效液相色谱法测定氨苄西林的生成量;根据《中国药典》(2000版,第338页至339页)规定的测定方法,采用高效液相色谱法测定阿莫西林的生成量。
实施例1 Z型头孢丙烯的制备
在100mL的三口反应瓶中,加入30mL水,而后再加入7-苯乙酰氨基-3-丙烯基(Z型)-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为丙烯基(Z型))0.5g,同时进行搅拌,滴加3M氨水调pH至6.5并使其溶解;然后向反应器中加入对羟基苯甘氨酸乙二醇酯2.1g,再加入PGA-450固定化酰化酶0.5g;使整个反应体系恒温在15~20℃,并用Metron自动滴加仪滴加4M盐酸,使整个反应体系pH保持在6.5~7.0;反应4小时后,取反应液进行HPLC测试(外标法),生成Z型头孢丙烯0.49g,总转化率为85.0%。
实施例2 E型头孢丙烯的制备
在100mL的三口反应瓶中,加入50mL水,而后再加入7-苯乙酰氨基-3-丙烯基(E型)-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为丙烯基(E型))0.5g,同时进行搅拌,滴加3M氨水调pH至7.5并使其溶解;然后向反应器中加入对羟基苯甘氨酸乙二醇酯2.5g,再加入PGA-450固定化酰化酶0.5g;使整个反应体系恒温在15~20℃,并用Metron自动滴加仪滴加4M盐酸,使整个反应体系pH保持在7.0~7.5;反应5小时后,取反应液进行HPLC测试(外标法),生成E型头孢丙烯0.45g,总转化率为78.1%。
实施例3 头孢氨苄的制备
在100mL的三口反应瓶中,加入25mL水,而后再加入7-苯乙酰氨基-3-甲基-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为甲基)0.5g,同时进行搅拌,滴加3M氨水调pH至8.0并使其溶解;然后向反应器中加入苯甘氨酸甲酯0.8g,再加入PGA-450固定化酰化酶0.5g;使整个反应体系恒温在10~15℃,并用Metron自动滴加仪滴加4M盐酸,使整个反应体系pH保持在5.5~6.0;反应8小时后,取反应液进行HPLC测试(外标法),生成头孢氨苄0.467g,总转化率为90.1%。
实施例4 头孢羟氨苄的制备
在100mL的三口反应瓶中,加入35mL水,而后再加入7-苯乙酰氨基-3-甲基-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为甲基)0.5g,同时进行搅拌,滴加三乙胺调pH至7.5并使其溶解;然后向反应器中加入对羟基苯甘氨酸甲酰胺0.9g,再加入PGA-450固定化酰化酶0.8g;使整个反应体系恒温在8~10℃,并用Metron自动滴加仪滴加4M盐酸,使整个反应体系pH保持在7.8~8.0;反应3小时后,取反应液进行HPLC测试(外标法),生成头孢羟氨苄0.48g,总转化率为92.2%。
实施例5 头孢克洛的制备
在100mL的三口反应瓶中,加入50mL水,而后再加入7-苯乙酰氨基-3-氯-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为氯原子)0.5g,同时进行搅拌,滴加20wt.%氢氧化钠调pH至8.0并使其溶解;然后向反应器中加入对羟基苯甘氨酸乙二醇酯2.6g,再加入IPA-750固定化酰化酶1.0g;使整个反应体系恒温在25~30℃,并用Metron自动滴加仪滴加15wt%硫酸,使整个反应体系pH保持在6.0~6.5;反应6小时后,取反应液进行HPLC测试(外标法),生成头孢克洛0.44g,总转化率为83.5%。
实施例6 头孢拉啶的制备
在100mL的三口反应瓶中,加入30mL水和10mL乙二醇,而后再加入7-苯乙酰氨基-3-甲基-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为甲基)0.5g,同时进行搅拌,滴加3M氨水调pH至6.5并使其溶解;然后向反应器中加入双氢苯甘氨酸乙二醇酯1.0g,再加入IPA-750固定化酰化酶0.5g;使整个反应体系恒温在15~20℃,并用Metron自动滴加仪滴加10wt%磷酸,使整个反应体系pH保持在5.5~6.0;反应7小时后,取反应液进行HPLC测试(外标法),生成头孢拉啶0.486g,总转化率为93.1%。
实施例7 Z型头孢丙烯的制备
在100mL的三口反应瓶中,加入10mL水,而后再加入7-苯氧乙酰氨基-3-丙烯基(Z型)-3-头孢菌素-4-羧酸(通式(III),R3为苯氧亚甲基,Z为丙烯基(Z型))0.5g,同时进行搅拌,滴加3M氨水调pH至7.5并使其溶解;然后向反应器中加入对羟基苯甘氨酸乙二醇酯1.9g,再加入IPA-450固定化酰化酶0.6g;使整个反应体系恒温在25~30℃,并用Metron自动滴加仪滴加4M盐酸,使整个反应体系pH保持在6.0~6.5;反应4小时后,取反应液进行HPLC测试(外标法),生成Z型头孢丙烯0.51g,总转化率为88.5%。
实施例8 头孢唑啉的制备
在100mL的三口反应瓶中,加入30mL水,而后再加入7-苯乙酰氨基-3-(2-甲基-1,3,4-噻二唑-5-基)硫亚甲基-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为(2-甲基-1,3,4-噻二唑-5-基)硫亚甲基))0.5g,同时进行搅拌,滴加3M氨水调pH至7.5并使其溶解;然后向反应器中加入1H-四氮唑乙酸乙二醇酯1.8g,再加入PGA-450固定化酰化酶2.0g;使整个反应体系恒温在5~10℃,并用Metron自动滴加仪滴加4M盐酸,使整个反应体系pH保持在6.5~7.0;反应7小时后,取反应液进行HPLC测试(外标法),生成头孢唑啉0.41g,总转化率为77.4%。
实施例9 Z型头孢丙烯的制备
在250mL的三口反应瓶中,加入100mL水,而后再加入7-苯乙酰氨基-3-丙烯基(Z型)-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为丙烯基(Z型))0.5g,同时进行搅拌,滴加5%氢氧化钠调pH至6.5并使其溶解;然后向反应器中加入对羟基苯甘氨酸乙二醇酯4.2g,再加入IPA-750固定化酰化酶3.5g;使整个反应体系恒温在15~20℃,并用Metron自动滴加仪滴加3M盐酸,使整个反应体系pH保持在5.0~5.5;反应16小时后,取反应液进行HPLC测试(外标法),生成Z型头孢丙烯0.41g,总转化率为71.1%。
实施例10 Z型头孢丙烯的制备
在150mL的三口反应瓶中,加入40mL水,而后再加入7-苯乙酰氨基-3-丙烯基(Z型)-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为丙烯基(Z型))0.5g,同时进行搅拌,滴加3M氨水调pH至6.5并使其溶解;然后向反应器中加入对羟基苯甘氨酸乙二醇酯1.8g,再加入PGA-450固定化酰化酶0.5g;使整个反应体系恒温在15~20℃,并用Metron自动滴加仪滴加3M盐酸,使整个反应体系pH保持在8.0~8.5;反应3小时后,取反应液进行HPLC测试(外标法),生成Z型头孢丙烯0.48g,总转化率为83.3%。
实施例11Z型头孢丙烯的制备
在100mL的三口反应瓶中,加入20mL水,而后再加入7-苯乙酰氨基-3-丙烯基(Z型)-3-头孢菌素-4-羧酸(通式(III),R3为苄基,Z为丙烯基(Z型))0.5g,同时进行搅拌,滴加4M氨水调pH至6.0并使其溶解;然后向反应器中加入对羟基苯甘氨酸乙二醇酯6.2g,再加入PGA-450固定化酰化酶4.5g;使整个反应体系恒温在15~20℃,并用Metron自动滴加仪滴加4M盐酸,使整个反应体系pH保持在7.0~7.5;反应2小时后,取反应液进行HPLC测试(外标法),生成Z型头孢丙烯0.53g,总转化率为91.9%。
实施例12 氨苄西林的制备
在150mL的三口反应瓶中,加入40mL水,而后再加入青霉素G钾(通式(IV),R3为苄基,)0.5g,同时进行搅拌,滴加5wt%氢氧化钠调pH至6.5并使其溶解;然后向反应器中加入苯甘氨酸甲酯3.5g,再加入IPA-750固定化酰化酶2.5g;使整个反应体系恒温在35~40℃,并用Metron自动滴加仪滴加4M盐酸使整个反应体系pH保持在6.0~6.2;反应6小时后,取反应液进行HPLC测试(外标法),生成氨苄西林0.45g,总转化率为90.0%。
实施例13 阿莫西林的制备
在150mL的三口反应瓶中,加入60mL水,而后再加入青霉素G钾(通式(IV),R3为苄基,)0.5g,同时进行搅拌,滴加4M氨水调pH至5.5并使其溶解;然后向反应器中加入羟基苯甘氨酸乙二醇酯2.2g,再加入PGA-450固定化酰化酶1.5g;使整个反应体系恒温在5~10℃,并用Metron自动滴加仪滴加4M盐酸使整个反应体系pH保持在5.5~6.0;反应8小时后,取反应液进行HPLC测试(外标法),生成阿莫西林0.41g,总转化率为82.0%。
实施例14 阿莫西林的制备
在150mL的三口反应瓶中,加入60mL水,而后再加入青霉素G钾(通式(IV),R3为苄基,)0.5g,同时进行搅拌,滴加4M氨水调pH至7.5并使其溶解;然后向反应器中加入对羟基苯甘氨酸乙二醇酯2.2g,再加入PGA-450固定化酰化酶1.5g;使整个反应体系恒温在25~30℃,并用Metron自动滴加仪滴加4M盐酸使整个反应体系pH保持在7.5~8.0;反应2小时后,取反应液进行HPLC测试(外标法),生成阿莫西林0.43g,总转化率为86.0%。
实施例15 氨苄西林的制备
在150mL的三口反应瓶中,加入40mL水,而后再加入青霉素G钾(通式(IV),R3为苄基,)0.5g,同时进行搅拌,滴加4M氨水调pH至6.5并使其溶解;然后向反应器中加入苯甘氨酸乙二醇酯3.0g,再加入PGA-450固定化酰化酶1.5g;使整个反应体系恒温在15~20℃,并用Metron自动滴加仪滴加4M盐酸使整个反应体系pH保持在6.0~6.2;反应6小时后,取反应液进行HPLC测试(外标法),生成氨苄西林0.42g,总转化率为84.0%。
酶催化“一锅法”制备β-内酰胺抗生素的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。





动态评分
0.0