

IPC分类号 : C12Q1/68,C12Q1/04,C12N15/10,C12N15/63,C12R1/00







专利摘要
一种生物技术领域的基于DNA随机改组技术的扩增内标制备方法,步骤包括:根据目的基因设计得到特异性检测引物和TaqMan探针;根据目标片断上的探针结合部位的DNA序列,采用DNA随机改组软件随机生成改组序列,将生成的改组序列分别替代目标片断上的探针结合部位的DNA序列,产生相应的待筛选的扩增内标序列;利用荧光探针设计软件和BLAST-N软件比对筛选扩增内标序列,得到软件评价分值最高且与其它致病微生物基因组序列非同源的序列,以此序列作为扩增内标序列;将步骤三得到的扩增内标序列克隆到载体上,构建含扩增内标序列的载体;检测得到的扩增内标序列。本发明有助于检测时显示抑制现象的存在,提高检测结果的准确率。
权利要求
1、一种基于DNA随机改组技术的扩增内标制备方法,其特征在于,包括如下步骤:
步骤一,根据目的基因设计得到特异性检测引物和TaqMan探针;
步骤二,根据目标片断上的探针结合部位的DNA序列,采用DNA随机改组软件随机生成改组序列,将生成的改组序列分别替代目标片断上的探针结合部位的DNA序列,产生相应的待筛选的扩增内标序列;
步骤三,利用荧光探针设计软件和BLAST-N软件比对筛选扩增内标序列,得到软件评价分值最高且与其它致病微生物基因组序列非同源的序列,以此序列作为扩增内标序列;
步骤四,将步骤三得到的扩增内标序列克隆到载体上,构建含扩增内标序列的载体;
步骤五,检测得到的扩增内标序列。
2、根据权利要求1所述的基于DNA随机改组技术的扩增内标制备方法,其特征是,步骤一中,所述特异性检测引物和TaqMan探针具体为,通过Genbank中公用的软件,选出单核细胞增生李斯特菌hlyA基因中序列特异性较高的序列区段,根据该区段序列,检测引物和TaqMan探针。
3、根据权利要求1所述的基于DNA随机改组技术的扩增内标制备方法,其特征是,步骤五中,所述检测具体为,在荧光定量PCR体系中分别添加梯度稀释的扩增内标和目标序列模板,计算扩增内标和目标序列的扩增效率,比较扩增内标与目标序列的扩增效率之差。
说明书
技术领域技术领域
本发明涉及一种生物技术领域的扩增内标制备方法,具体是一种基于DNA随机改组技术的扩增内标制备方法。
技术背景背景技术
荧光定量PCR技术是在PCR定性技术基础上发展起来的核酸定量技术,该技术在PCR反应体系中加入荧光标记探针,利用荧光信号的积累实时监测整个PCR进程,并根据荧光信号推断目的基因的初始量。荧光定量PCR技术与以前的以终点法进行定量的PCR技术相比具有很大的优势。首先,它不仅操作简便、快速高效,高通量,能实现多重反应,而且具有很高的灵敏性、重复性和特异性。其次,由于是在封闭的体系中完成扩增并进行实时测定,大大降低了污染的可能性并且无需在扩增后进行操作。目前,荧光定量PCR技术作为一个极有效的实验技术,已被广泛地应用于分子生物学研究的各个领域。
但是荧光定量PCR方法在不同的实验室或检测部门所检测的目的基因和操作流程有一定的差异,没有形成统一标准,得到的检测结果也不尽相同。近年来的实践应用表明,增菌培养和DNA提取过程中存在的抑制成分可影响荧光定量PCR反应的扩增效率,干扰检测结果的准确性。尽管荧光定量PCR方法在不断的改进和完善,却不能有效地解决试验过程中抑制成分对PCR扩增效率的影响。为了解决荧光定量PCR方法中普遍存在的抑制现象问题,近几年在荧光定量PCR扩增体系中引入了扩增内标,其主要原理是:在荧光定量PCR中使用与目的基因相似的扩增内标,它的两端引物结合序列与目的基因完全一致,但具有不同的探针结合序列,使之不能与目的基因的探针结合,只能与内标探针结合。于是,在含内标的荧光定量PCR体系中有两种DNA模板——目的基因片段和扩增内标;一对引物,该对引物既能与目标基因片段结合,也与扩增内标结合;两条探针——目标基因探针和扩增内标探针,分别结合目标基因片段和扩增内标。在扩增内标与目标片断的共同扩增中,通过扩增内标的扩增效率来反应目标片断的扩增效率,从而达到指示抑制现象和准确定量的目的。
荧光定量PCR反应中的扩增效率主要受引物序列及扩增片断的长度和GC含量的影响。目前,扩增内标序列往往都是根据传统经验获得,缺乏具体有效的扩增内标设计方法,很难保证扩增内标与目标片断序列长度和GC含量的一致性,从而导致两者扩增效率的不同,进而影响到最终检测结果的准确性。
近年来随着计算机技术的飞速发展,许多的计算机程序被运用到生物技术领域,计算机随机改组技术便是其中一员。Jacka S等在《Stochastic Processesand Their Applications》(随机过程及其应用)2007年117期708-719页发表了题为“Random orderings of the integers and card shuffling”(整数和纸牌的随机改组)的论文,文中评述了基于计算机程序的随机改组方法,该方法使得基于计算机程序的DNA序列定向筛选成为可能,它能够根据指定序列随机生成大量的改组序列,这些序列与原始序列具有相同的长度和GC含量。
经对现有技术的文献检索发现,尚未见有关基于DNA随机改组技术的扩增内标制备方法的报道。
发明内容发明内容
本发明的目的在于提供一种基于DNA随机改组技术的扩增内标制备方法。本发明可保证扩增内标序列与目标序列具有相同的扩增效率,有助于检测时显示抑制现象的存在,提高检测结果的准确率,为满足临床和检疫执法过程中所急需的对致病微生物调查和检测提供有效可靠的技术手段。
本发明通过以下技术方案实现,本发明涉及一种基于DNA随机改组技术的扩增内标制备方法,包括如下步骤:
步骤一,根据目的基因设计得到特异性检测引物和TaqMan探针;
步骤二,根据目标片断上的探针结合部位的DNA序列,采用DNA随机改组软件随机生成改组序列,将生成的改组序列分别替代目标片断上的探针结合部位的DNA序列,产生相应的待筛选的扩增内标序列;
步骤三,利用荧光探针设计软件和BLAST-N软件比对筛选扩增内标序列,得到软件评价分值最高且与其它致病微生物基因组序列非同源的序列,以此序列作为扩增内标序列;
步骤四,将步骤三得到的扩增内标序列克隆到载体上,构建含扩增内标序列的载体;
步骤五,检测得到的扩增内标序列。
步骤一中,所述特异性检测引物和TaqMan探针具体为,通过Genbank中公用的BLAST软件,选出单核细胞增生李斯特菌hlyA基因中序列特异性较高的序列区段,根据该区段序列,利用软件Bacon Designer 5.0设计特异性检测引物和TaqMan探针。
步骤五中,所述检测具体为,在荧光定量PCR体系中分别添加梯度稀释的扩增内标和目标序列模板,计算扩增内标和目标序列的扩增效率,比较扩增内标与目标序列的扩增效率之差。
本发明利用DNA随机改组技术,根据目标片断的序列特征,随机生成改组序列,然后通过荧光探针设计软件分析和BLAST-N比对筛选,可找到一条理想的内标序列。本发明得到的扩增内标序列与目标序列具有相同的长度和GC含量,从而保证两者扩增效率的一致性,该扩增内标有助于检测荧光定量PCR反应体系中存在的抑制现象,提高检测结果的准确率。
本发明具有如下的有益效果:可通过本发明的扩增内标的扩增效率来指示目标片断的扩增效率,从而实现精确定量;本发明有助于显示荧光定量PCR检测体系中抑制现象,克服了因增菌培养和DNA提取过程中存在的抑制成分对PCR扩增效率的影响,提高了荧光定量PCR检测的准确率,为临床和检疫执法过程中所急需的对致病微生物调查和检测提供可靠的技术手段。
本发明中所涉及的菌株单核细胞增生李斯特菌和大肠杆菌DH5α已在《龙飞,朱欣娜,张忠明,史贤明;基于活菌内标的单核细胞增生李斯特菌荧光定量PCR方法的建立,疾病感染诊断微生物学,2008,62(4):374-381》文献中公开。
附图说明附图说明
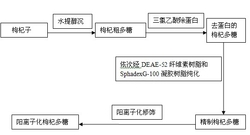
图1为扩增内标序列设计步骤示意图;
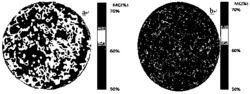
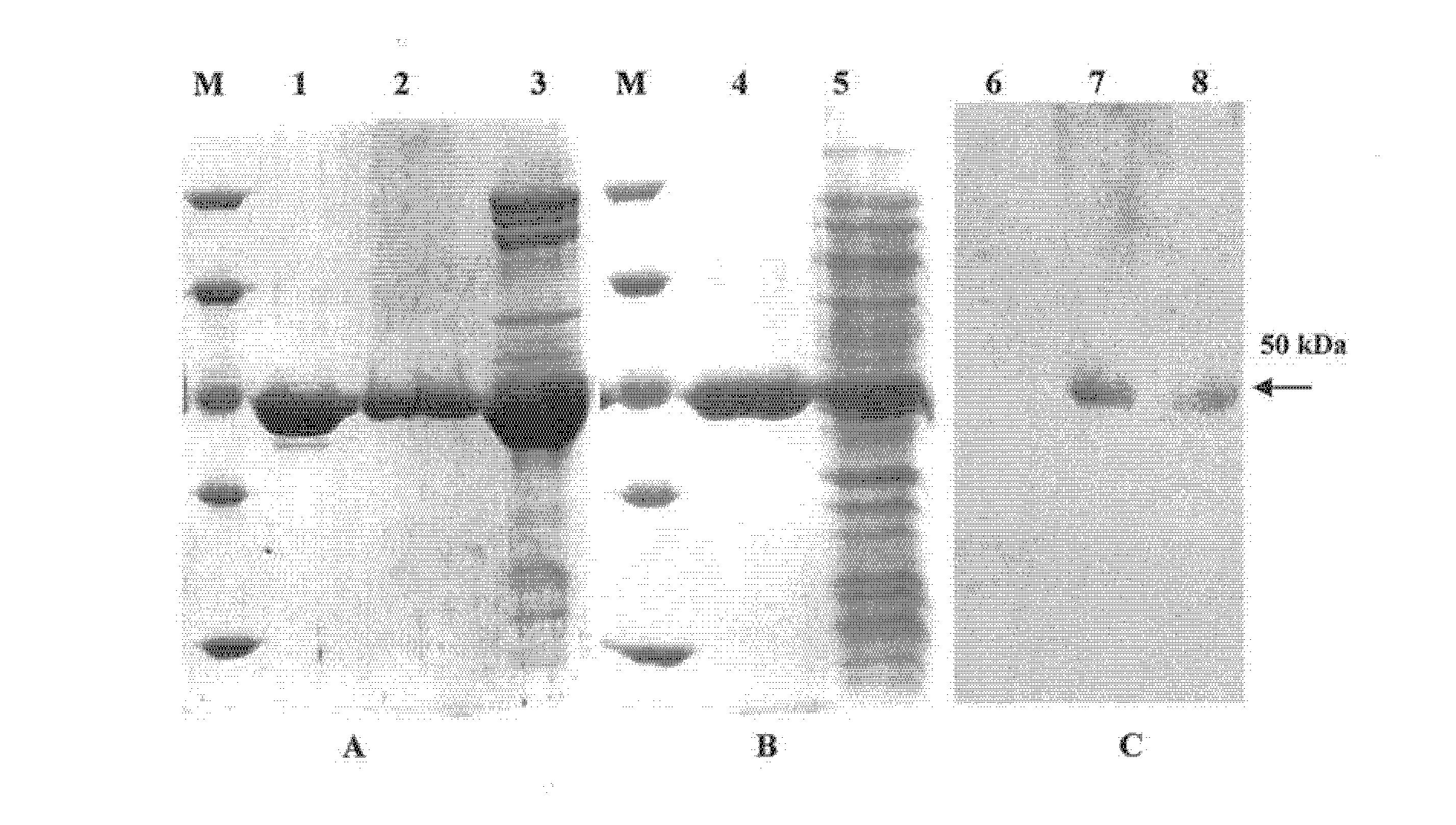
图2为大肠杆菌DH5α阳性克隆的PCR检测结果示意图;
图3为目标片段和扩增内标序列扩增效率评价结果示意图。
具体实施方式具体实施方式
下面对本发明的实施例作详细说明:本实施例在以本发明技术方案为前提下进行实施,下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如Sambrook等分子克隆:实验室手册(New York:Cold Spring Harbor LaboratoryPress,1989)中所述的条件,或按照制造厂商所建议的条件。本发明给出了详细的实施方式和具体的操作过程,但保护范围不限于下述的实施例。
实施例
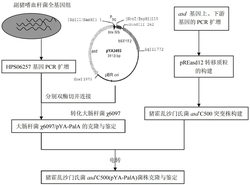
图1为扩增内标序列设计步骤示意图,图中:(a)为目标片段探针的设计;(b)为目标片段探针结合部位序列的随机改组;(c)为待筛选扩增内标片段的生成;(d)为扩增内标片段的筛选;(e)为扩增内标序列的确定。
步骤一,分析目的基因,通过设计得到特异性检测引物和TaqMan探针
对单核细胞增生李斯特菌的已知特异基因进行分析,从中选出用于检测的目的基因hlyA。通过Genbank中公用的BLAST软件,将hlyA基因的序列分别与其他微生物进行比对,选出特异性较高的序列区段,根据这段序列,利用软件Bacon Designer 5.0设计一对引物与探针。
引物及探针序列如下:
单核细胞增生李斯特菌(hlyPF/hlyPR、probe-W)
hlyPF:5’-CATGGCACCACCAGCATC-3’
hlyPR:5’-CATCCGCGTGTTTCTTTTCG-3’
probe-W:5’FAM-CCGCCTGCAAGTCCTAAGACGCCA-ECLIPCE-3’。
(a)检测目的基因的序列特征:
*长度:65碱基对
*类型:核酸
*链型:双链
*拓扑结构:线形
(b)分子类型:DNA
(c)最初来源:单核细胞增生李斯特菌
(d)序列描述:
CATGGCACCACCAGCATCTCCGCCTGCAAGTCCTAAGACGCCAATCGAAAAGAAACACGCGGATG。
步骤二,分析目标片断上的探针结合部位的DNA序列,采用DNA随机改组软件随机生成改组序列,将生成的改组序列分别替代目标片断上的探针结合部位的DNA序列,产生相应的待筛选的扩增内标序列
①扩增内标随机改组序列的生成
根据目标片断上的探针结合部位的DNA序列,采用DNA随机改组软件BioToolKit 320随机生成n组(n≥1000)改组序列;
②待筛选扩增内标序列的生成
将n组(n≥1000)改组序列分别替代目标片断上的探针结合部位的DNA序列,产生n组(n≥1000)待筛选的扩增内标序列。
步骤三,利用荧光探针设计软件和BLAST-N比对筛选扩增内标序列,得到软件评价分值最高且与其它致病微生物基因组序列非同源的序列,以此序列作为扩增内标序列
通过荧光探针设计软件Bacon Designer 5.0分析找到软件评价分值最高的4-10组序列,然后通过BLAST-N软件比对,选取一组软件评分最高且与其它致病微生物基因组序列非同源的序列,以此作为扩增内标序列并设计相应的检测探针。
相应探针及扩增内标序列如下:
probe-I:5’-HEX-ATAGGAGCACTCGCCGCCCACATC-ECLIPCE-3’
(a)扩增内标的序列特征:
*长度:65碱基对
*类型:核酸
*链型:双链
*拓扑结构:线形
(b)分子类型:DNA
(c)最初来源:人工合成
(d)序列描述:
CATGGCACCACCAGCATCTATAGGAGCACTCGCCGCCCACATCATCGAAAAGAAACACGCGGATG
步骤四,将步骤三得到的扩增内标序列克隆到载体上,构建含扩增内标序列的载体
①利用单核细胞增生李斯特菌特异性引物(hlyPF/hlyPR)对内标序列进行PCR扩增;
②PCR扩增产物片段与载体在T4DNA连接酶和酶连缓存液LigationSolution I作用下16℃过夜;
③转化大肠杆菌DH5α,用氯化钙法转化大肠杆菌DH5α,然后涂布于选择性平板上,选择性平板具体为:90mm培养皿,100mg/ml氨苄青霉素10μl,20%的IPTG溶液7μl,2%的X-gal溶液40μl,37℃培养12h;用灭菌的牙签从选择性平板上挑取白色菌落,接菌到装有5ml LB培养液的PA瓶中,再在37℃下以150r/min培养8h,提取克隆载体;
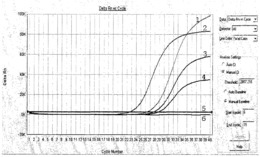
④检测,利用扩增内标特异引物和探针(hlyPF和hlyPR、probe-I)对筛选的大肠杆菌阳性克隆进行荧光PCR检测,具体结果如图2,图2中:曲线1,以扩增内标引物和探针(hlyPF/hlyPR、probe-W)扩增的结果;曲线2,阴性对照;对PCR检测所确定的大肠杆菌阳性克隆抽取克隆载体进行进一步的测序验证。
步骤五,检测得到的扩增内标序列
①提取单核细胞增生李斯特菌基因组DNA并测定相应含量,经10倍梯度稀释后备用,最终荧光定量PCR反应中加入的目标片段模板量分别从2.52×107fg到25.2fg;
②提取含扩增内标的克隆载体并测定相应含量,经10倍梯度稀释后备用,最终荧光定量PCR反应中加入的扩增内标模板量分别从2.4×107fg到24fg,确定的反应体系如表1。
表1
PCR循环参数为:45个循环,每个循环的程序包括95℃变性5s,退火延伸温度65℃,时间为20s,循环结束后降温至4℃,结束所有操作程序。
③在荧光定量PCR体系中分别添加梯度稀释的扩增内标和目标序列模板,计算扩增内标和目标序列的扩增效率,比较扩增内标与目标序列的扩增效率之差,如果差值≦0.1,则说明该扩增内标是可用的;
扩增效率的计算可以采用梯度稀释法,将稀释后模板浓度的对数值与所得阈值循环数值(threshold cycle,CT)做图,在一定范围内应得到一条线性回归直线,利用公式E=10-1/K—1(E为扩增效率,K为该直线斜率)即可计算出E值。将目标片段和扩增内标序列稀释后浓度的对数值与所得阈值循环数值分别做图,在一定范围内通过线性回归得到一条直线,线性回归结果如图3所示:图3中,1:以梯度稀释目标片段DNA为模板,计算所得线性回顾方程为y=-3.2179x+40.354,方程斜率K=-3.2179,扩增效率E=1.05;2:以梯度稀释扩增内标DNA为模板,计算所得线性回顾方程为y=-3.1817x+40.328,方程斜率K=-3.1817,扩增效率E=1.06,比较目标片段和扩增内标序列扩增效率值可知两者两者之差<0.1,具有非常相似的扩增效率。
本实施例可广泛运用于荧光定量PCR扩增内标序列的制备,所制备的扩增内标序列与其它致病微生物基因组非同源,且与目标序列具有相同的长度和GC含量,保证了目标片段和扩增内标序列扩增效率的一致性;本实施例有助于检测时显示抑制现象的存在,提高检测结果的准确率,能够满足临床和检疫执法过程中对致病微生物调查和检测。
基于DNA随机改组技术的扩增内标制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












动态评分
0.0