







专利摘要
本发明公开一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法。该方法首先通过MaterialsStudio软件构建氧化石墨烯分子与聚碳化二亚胺分子的混合结构,然后通过能量最小化和分子动力学运算,利用xLink交联程序进行结构交联,最终得到交联结构模型。本发明的模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法具有模拟时间短的特点,所有运算均可并行计算,视并行核数和体系大小不同,最快几个小时便可完成,从而为复杂交联体系的计算模拟提供了一种可靠的结构建模方法。
权利要求
1.一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,其特征在于具体步骤包括:
(1)、利用MS软件中的Visualizer模块分别构建氧化石墨烯分子、聚碳化二亚胺分子的结构A1、B1;
(2)、利用MS软件中的Forcite模块进行能量最小化运算;
利用MS软件中的Forcite模块分别对步骤(1)所得的两个结构A1、B1进行能量最小化运算,在Forcite模块中选用Smart Minimize方法、COMPASS分子力场、Atom based非键作用求和方法,分别得到能量最优的结构A2、B2;
(3)、利用MS软件中的Amorphous Cell模块进行结构混合运算;
利用MS软件中的Amorphous Cell模块对步骤(2)所得的两个结构A2、B2进行结构混合运算,其中分子力场选用COMPASS力场,库伦作用求和选用Ewald方法,范德华作用求和选用Atom based方法,得到混合结构C1;
混合结构运算中,氧化石墨烯分子、聚碳化二亚胺分子的加载数至少各设为1个,控制混合结构中总原子数不超过5000个原子;
(4)、利用MS软件中的Forcite模块进行能量最小化和分子动力学弛豫的运算;
利用MS软件中的Forcite模块对步骤(3)所得的混合结构文件C1进行能量最小化和分子动力学弛豫的运算;
其中进行能量最小化运算时,选用Smart Minimize方法、 COMPASS分子力场、Ewald非键作用求和方法;
进行分子动力学弛豫运算时,选用COMPASS分子力场、NPT系综、Ewald非键作用求和方法,温度选用200-500K,压力选用1大气压即1.0×10
(5)、利用xLink程序“虚拟交联反应”运算;
对步骤(4)运算所得的结构进行“虚拟交联反应”运算,步骤如下:
交联反应距离初始值设定为
利用MS软件Visualizer模块的Measure工具测量步骤(4)运算所得的结构中任意一对C-N原子对中C、N原子之间的距离D;
其中所述的C-N原子对是指由氧化石墨烯分子中羰基基团中的C原子与聚碳化二亚胺分子中N=C=N基团中的N原子形成的原子对;
当C、N原子之间的距离D小于或等于当前的交联反应距离时,发生交联反应,构建交联结构;
所述的构建交联结构,即利用MS软件Visualizer模块将上述发生交联反应的结构中对应的羰基基团中的C原子与 N=C=N基团中的N原子直接形成单键,将羰基基团中的C原子上羟基基团中的O原子与N=C=N基团中的C原子直接形成双键,然后根据各原子配位情况进行H原子饱和;
当C、N原子之间的距离D大于当前的交联反应距离时,不发生交联反应,不进行操作;
再对另外一对C-N原子对运行本步骤(5),直至步骤(4)运算所得的结构中所有的C-N原子对均已判断是否发生交联反应,且所有符合上述发生交联反应条件的C-N原子对均已构建交联结构,得到交联结构D1;
(6)、利用MS软件中的Forcite模块进行温度循环的分子动力学弛豫运算;
利用MS软件中的Forcite模块对步骤(5)“虚拟交联反应”运算所得的交联结构D1进行温度循环的分子动力学弛豫运算,Forcite模块中力场选用COMPASS分子力场,系综选用NVT系综,温度循环范围选用300K-(500-800K)-300K,温度变化量选用50-100K,每个温度下分子动力学运算的总模拟时间均选用300-1000ps;
每次分子动力学运算均以上一个分子动力学运算后的结果作为初始结构,每次分子动力学运算的总模拟时间均相同,直至以上温度由300K升温到500-800K后再降温到300K的分子动力学弛豫运算完成,结束温度循环的分子动力学弛豫运算,计算交联结构D1的反应度;
所述交联结构D1的反应度为交联结构D1中所用的氧化石墨烯分子中已经发生交联反应的羰基基团的个数与交联结构D1中所用的氧化石墨烯分子中所有羰基基团个数的比值;
(7)、步骤(6)运算所得交联结构D1的反应度与预先设定的反应度的目标值进行比较;
所述反应度的目标值,根据实际需要在步骤(5)的虚拟交联反应程序中进行预先设定,最大值为100%;
当步骤(6)运算所得交联结构D1的反应度达到预先设定的反应度的目标值,此时的交联结构D1即为满足要求的氧化石墨烯与聚碳化二亚胺交联结构;
当步骤(6)运算所得交联结构D1的反应度未达到预先设定的反应度的目标值,且当前交联反应距离小于
当步骤(6)运算所得交联结构D1的反应度未达到预先设定的反应度的目标值,且当前交联反应距离达到交联反应距离最大值
(8)、利用MS软件Visualizer模块的Measure工具测量并统计,步骤(7)中当交联结构D1的反应度达到预先设定的反应度的目标值时交联结构D1中的C-N键的键长L,以判断整个构建过程是否结束:
当
当
当
2.如权利要求1所述的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,其特征在于步骤(3)中结构混合运算中,氧化石墨烯分子加载数为1-10个,聚碳化二亚胺分子加载数为5-20个。
3.如权利要求1或2所述的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,其特征在于:
步骤(4)中温度选用300-400K,总模拟时间选用800-1000ps;
步骤(5)中交联反应距离初始设定值为
步骤(5)中在后续步骤(7)的运算中,要增大交联反应距离时,每次增大
步骤(6)中温度循环范围选用300K-600K-300K,温度变化量选用50-75K,每个温度下分子动力学运算的总模拟时间相同,均为500-800ps。
4.如权利要求1或2所述的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,其特征在于:
步骤(3)中混合结构运算中,氧化石墨烯分子的加载数为10个、聚碳化二亚胺分子的加载数为20个;
步骤(4)中温度选用400K,总模拟时间选用800ps;
步骤(5)中交联反应距离初始设定值为
步骤(5)中在后续步骤(7)的运算中,要增大交联反应距离时,每次增大
步骤(6)中温度循环范围选用300K-620K-300K,温度变化量选用80K,每个温度下分子动力学运算的总模拟时间相同,均为800ps。
5.如权利要求1或2所述的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,其特征在于:
步骤(3)中混合结构运算中,氧化石墨烯分子的加载数为1个、聚碳化二亚胺分子的加载数为10个;
步骤(4)所述的温度为300K,总模拟时间为1000ps;
步骤(5)中交联反应距离初始值设定为
步骤(5)中在后续步骤(7)的运算中,要增大交联反应距离时,每次增大
步骤(6)中温度循环范围选用300K-600K-300K,温度变化量选用75K,每个温度下分子动力学运算的总模拟时间均选用500ps。
6.如权利要求1或2所述的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,其特征在于:
步骤(3)中混合结构运算中,氧化石墨烯分子的加载数为2个、聚碳化二亚胺分子的加载数为5个;
步骤(4)所述的温度为200K,总模拟时间为500ps;
步骤(5)中交联反应距离初始值设定为
步骤(5)中在后续步骤(7)的运算中,要增大交联反应距离时,每次增大
步骤(6)中温度循环范围选用300K-500K-300K,温度变化量选用50K,每个温度下分子动力学运算的总模拟时间均选用300ps。
7.如权利要求1或2所述的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,其特征在于:
步骤(3)中混合结构运算中,氧化石墨烯分子的加载数为5个、聚碳化二亚胺分子的加载数为10个;
步骤(4)所述的温度为500K,总模拟时间为1500ps;
步骤(5)中交联反应距离初始值设定为
步骤(5)中在后续步骤(7)的运算中,要增大交联反应距离时,每次增大
步骤(6)中温度循环范围选用300K-800K-300K,温度变化量选用100K,每个温度下分子动力学运算的总模拟时间均选用1000ps。
8.如权利要求1所述的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法适用于由羰基基团与N=C=N基团发生交联反应形成的复杂交联结构模型的构建。
说明书
技术领域
本发明涉及一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,属于交联聚合物领域。
背景技术
腐蚀是冶金、交通运输等工业行业领域中的重大挑战之一。使用聚合物防腐材料是有效的防腐手段之一。聚合物若通过交联反应形成三维网络状交联结构,则其力学性能、热稳定性、耐磨性、耐溶剂性及抗蠕变性都有不同程度的提高。具有二维结构的石墨烯由于具有优异的性能引起了广泛关注。石墨烯化学性质稳定,具有优异的阻隔性能,因此可分散到聚合物防腐材料中形成复合涂层,具有极大的防腐应用潜力。在实际应用中,因分散操作的难易程度,实际使用的是氧化石墨烯。氧化石墨烯的结构中通常含有羟基、羧基等亲水基团,因此更易于分散。
氧化石墨烯若通过化学作用尤其交联反应分散到聚合物中,不但可以增强复合材料的防腐性能,而且可以增强复合材料的力学性质、热稳定性等。
实验上可通过盐雾实验测试氧化石墨烯-聚合物复合材料的防腐性能,但无法表征氧化石墨烯分散、交联等结构特征对防腐性能的影响,而利用计算机模拟手段可弥补这一不足。通过计算机模拟不但可以直观地观察氧化石墨烯的分散和交联情况,而且可以定量计算腐蚀介质在氧化石墨烯-聚合物复合材料中的扩散系数,直接用于比较防腐性能的好坏,还可以计算氧化石墨烯-聚合物复合材料的弹性模量等力学性质。利用计算机模拟构建结构模型的方法不但可以节省实验成本,而且可以从微观层面更好地解释实验现象和材料性能。
到目前为止,虽有文献报道氧化石墨烯接枝不同的官能团或长链分子的研究,但通过计算机模拟构建氧化石墨烯与聚合物形成复杂交联体系结构的研究未见报道。理论上,可以基于第一性原理分子动力学或反应分子动力学方法通过计算机模拟交联反应过程从而构建复杂交联体系的结构,但是存在模拟时间长、反应度不可控等技术问题。
而本发明正是为了解决上述复杂交联体系结构难以构建、交联反应过程模拟时间长、反应度不可控等技术问题而提供的一种计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构的方法,该模拟构建方法,由于采用以指定原子之间的距离为主要变量的“虚拟交联反应”技术,因此具有操作简单、模拟时间短、反应度可控等优点,可以获得比现有技术更好的复杂交联结构。
Materials Studio软件全尺度材料模拟平台,不仅拥有优异的操作界面,快捷实现模型搭建、参数设定以及结果的可视化分析,而且融合多种模拟方法,整合多个功能模块,实现从电子结构解析到宏观性能预测的全尺度科学研究,多种应用程序接口以及脚本编写功能的添加使其能够更好的满足各类用户的研究需求。
发明内容
本发明的目的是为了解决上述的复杂交联结构模型难以构建、交联反应过程模拟时间长、反应度不可控等技术问题而提供一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,该方法具有操作简单、模拟时间短、反应度可控的优点。
本发明的技术原理
本发明主要涉及Materials Studio软件(以下简称MS软件,版本不限,优选8.0及以上版本)中的Visualizer、Forcite、Amorphous Cell模块和COMPASS分子力场。
Visualizer模块是Materials Studio软件的图形化界面,可用于搭建、调整各类三维可视的结构模型,提供模块参数设置、结果分析的视窗界面;提供结构文件、参数文件以及结果文件的管理界面;提供计算进程的监控界面;对模拟结果进行各种分析,可与结构模型相结合进行数据的二维、三维显示,可以给出数据的图表,可对特定的结果进行动画演示;支持多种结构、图形、文本文件格式的输入和输出;支持不同功能模块间结构数据的共享;提供Perl语言环境。
Forcite模块是Materials Studio软件中的分子力学和分子动力学模拟程序,可以对分子、表面或三维周期性材料体系进行快速的能量计算、几何优化以及各种系综下的动力学模拟研究,可以分析材料体系的各种结构参数、热力学性质、力学性质、动力学性质以及统计学性质。主要应用于有机、无机小分子、有机金属络合物、高分子聚合物、纳米及多孔材料、部分金属、金属氧化物晶体及晶体表界面结构的研究。
Amorphous Cell模块是Materials Studio软件中采用蒙特卡洛方法搭建无定形模型的工具,可用于搭建具有多种组分及不同配比的高分子共混模型、溶液模型、复合材料模型、固液/固气界面模型、孔道填充模型、向列型液晶模型等,对塑料、玻璃、食品、化工以及复合材料等领域的模拟工作具有重要的辅助作用。
COMPASS分子力场是Materials Studio软件中基于量子力学方法,并且能够对凝聚态体系进行原子尺度模拟研究的分子力场,对其参数有效性的考察,不仅包括了单分子(气态)的量子力学计算结果以及实验结果,还充分考虑了其凝聚态性能。因此,COMPASS可在很大的温度、压力范围内,精确地预测多种单分子及其凝聚态的结构、构象、振动及热物理性质。
本发明的技术方案
一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,具体步骤包括:
(1)、利用MS软件中的Visualizer模块分别构建氧化石墨烯分子、聚碳化二亚胺分子的结构A1、B1;
(2)、利用MS软件中的Forcite模块进行能量最小化运算;
利用MS软件中的Forcite模块分别对步骤(1)所得的两个结构A1、B1进行能量最小化运算,在Forcite模块中选用Smart Minimize方法、COMPASS分子力场、Atom based非键作用求和方法,分别得到能量最优的结构A2、B2;
(3)、利用MS软件中的Amorphous Cell模块进行结构混合运算;
利用MS软件中的Amorphous Cell模块对步骤(2)所得的两个结构A2、B2进行混合结构运算,Amorphous Cell模块中分子力场选用COMPASS力场,库伦作用求和选用Ewald方法,范德华作用求和选用Atom based方法,得到混合结构C1;
混合结构运算中,氧化石墨烯分子、聚碳化二亚胺分子的加载数至少各设为1个,可根据需要增大各个分子的加载数,没有数目上限,但数目过多会影响整体运算速度,控制总原子数不超过5000个原子;
(4)、利用MS软件中的Forcite模块进行能量最小化和分子动力学弛豫运算;
利用MS软件中的Forcite模块对步骤(3)所得的混合结构C1进行能量最小化和分子动力学弛豫运算;
其中进行能量最小化运算时,选用Smart Minimize方法、COMPASS分子力场、Ewald非键作用求和方法;
进行分子动力学弛豫运算时,选用COMPASS分子力场、NPT系综、Ewald非键作用求和方法,温度选用200-500K,优选300-400K,压力选用1大气压即1.0×10
(5)、利用xLink程序“虚拟交联反应”运算;
对步骤(4)运算所得的结构进行“虚拟交联反应”运算,步骤如下:
交联反应距离初始值设定为 仅运行本步骤(5)时,此交联反应距离不做改变;经后续步骤(7)运算判断后,需要增大交联反应距离时,每次增大 交联反应距离的最大值设定为
利用MS软件Visualizer模块的Measure工具测量步骤(4)运算所得的结构中任意一对C-N原子对(所述的C-N原子对是指由氧化石墨烯分子中羰基基团中的C原子与聚碳化二亚胺分子中N=C=N基团中的N原子形成的原子对,后同)中C、N原子之间的距离D,判断此距离D是否小于或等于当前的交联反应距离,以此作为判断是否发生交联反应的依据;
当C、N原子之间的距离D小于或等于当前的交联反应距离时,发生交联反应,构建交联结构;
所述的构建交联结构,即利用MS软件Visualizer模块将上述发生交联反应的结构中对应的羰基基团中的C原子与N=C=N基团中的N原子直接形成单键,将羰基基团中的C原子上羟基基团中的O原子与N=C=N基团中的C原子直接形成双键,然后根据各原子配位情况进行H原子饱和;
当C、N原子之间的距离D大于当前的交联反应距离时,不发生交联反应,不进行操作;
再对另外一对C-N原子对运行本步骤(5),直至步骤(4)运算所得的结构中所有的C-N原子对均已判断是否发生交联反应,且所有符合上述发生交联反应条件的C-N原子对均已构建交联结构,得到交联结构D1;
(6)、利用MS软件中的Forcite模块进行温度循环的分子动力学弛豫运算;
利用MS软件中的Forcite模块对步骤(5)“虚拟交联反应”运算所得的交联结构D1进行温度循环的分子动力学弛豫运算,Forcite模块中力场选用COMPASS分子力场,系综选用NVT系综,温度循环范围选用300K-(500-800K)-300K,温度变化量选用50-100K,每个温度下分子动力学运算的总模拟时间均选用300-1000ps;
每次分子动力学运算均以上一个分子动力学运算后的结果作为初始结构,每次分子动力学运算的总模拟时间均相同,直至以上温度由300K升温到500-800K后再降温到300K的分子动力学弛豫运算完成,结束温度循环的分子动力学弛豫运算,计算交联结构D1的反应度;
所述交联结构D1的反应度为交联结构D1中所用的氧化石墨烯分子中已经发生交联反应的羰基基团的个数与交联结构D1中所用的氧化石墨烯分子中所有羰基基团个数的比值;
以温度循环范围为300K-800K-300K、温度变化量为100K为例,即首先在温度300K条件下进行分子动力学弛豫运算,然后依次进行温度400、500、600、700、800K条件下的分子动力学弛豫运算,运算完成后再依次进行700、600、500、400、300K条件下的分子动力学弛豫运算;
(7)、步骤(6)运算所得交联结构D1的反应度与预先设定的反应度的目标值进行比较;
所述反应度的目标值,是根据实际需要在步骤(5)的虚拟交联反应程序中预先设定的,预先设定的反应度的目标值最大值为100%;
当步骤(6)运算所得的交联结构D1的反应度达到预先设定的反应度的目标值,此时的交联结构D1即为满足要求的氧化石墨烯与聚碳化二亚胺交联结构;
当步骤(6)运算所得的交联结构D1的反应度未达到预先设定的反应度的目标值,且当前交联反应距离小于 则将当前交联反应距离增大 然后重新进行执行步骤(5)-(7),直至交联结构D1的反应度达到预先设定的反应度的目标值;
当步骤(6)运算所得的交联结构D1的反应度未达到预先设定的反应度的目标值,且当前交联反应距离达到交联反应距离最大值 运算暂停,提示交联结构D1的反应度未达到预先设定的反应度的目标值,重新进行步骤(3)-(7),直至交联结构D1的反应度达到预先设定的反应度的目标值;
(8)、步骤(7)中所得的交联结构D1的反应度达到预先设定的反应度的目标值后,利用MS软件Visualizer模块的Measure工具测量并统计所得的交联结构D1中的C-N键的键长L,以判断整个构建过程是否结束:
当 时,构建的交联结构D1合理,未出现穿环等异常结构,整个构建过程结束;
当 时,可能出现结构未能完全弛豫的情况,此时需要重新运行步骤(6)-(8),直至 整个构建过程结束;
当 时,可能出现穿环等不合理结构,此时需要运行步骤(3)-(8),直至 整个构建过程结束。
上述的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,由于核心部分步骤(5)“虚拟交联反应”运算仅涉及羰基基团和N=C=N基团,对基团周围化学环境并无要求和限制,因此适用于任意的由羰基基团与N=C=N基团发生交联反应形成复杂交联结构模型的构建。在本发明原理基础上通过适当修改,还可推广至任意类型交联反应形成的交联结构的构建。
本发明的有益技术效果
本发明的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,具有操作简单的特点,能量最小化和分子动力学运算均有图形界面,可以通过下拉菜单中选择的方式设置参数,“虚拟交联反应”运算只需在脚本中设置关键参数即可;所有参数均有默认值。
进一步,本发明的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,具有模拟时间短的特点,所有运算均可并行计算,视并行核数和体系大小不同,最快几个小时便可完成。
进一步,本发明的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,具有反应度可控的特点,反应度作为主要目标参数可以在“虚拟交联反应”运算中直接设定,只有达到设定的(目标)反应度,构建过程运算才基本结束。
进一步,本发明的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,具有组分范围大的特点,氧化石墨烯分子加载数、氧化石墨烯分子中的羰基基团数目、聚碳化二亚胺分子加载数、聚碳化二亚胺分子中N=C=N基团的数目均无限制,可以任意比例组合,此点在不利用计算机进行模拟构建的实验上是无法实现的。
进一步,本发明的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,具有适用体系广的特点,可直接用于任意由羰基基团与N=C=N基团发生交联反应形成复杂交联结构的模型构建。在本发明原理基础上通过适当修改,还可推广至任意类型交联反应形成的交联结构的构建。
进一步,本发明的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,具有计算环境灵活的特点,既可以在Windows版本的MS软件中进行,也可以在Linux版本的MS软件中进行;既可以通过图形界面提交运算,也可以通过命令形式提交运算;硬件既可以是普通PC机,也可以是服务器或集群。
附图说明
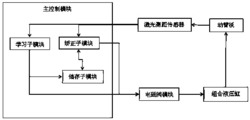
图1、氧化石墨烯与聚碳化二亚胺交联结构的构建过程的逻辑框图;
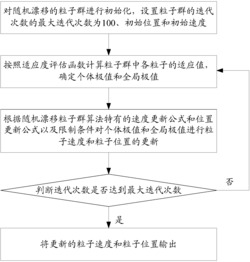
图2、xLink程序“虚拟交联反应”运算的逻辑框图;
图3、实施例1中利用计算机模拟构建2个氧化石墨烯分子和5个聚碳化二亚胺分子交联结构模型中部分交联结构的示意图;
图4、实施例1中利用计算机模拟构建2个氧化石墨烯分子和5个聚碳化二亚胺分子交联结构模型中交联反应形成的C-N键的键长在不同长度的分布概率图;
图5、实施例2中利用计算机模拟构建5个氧化石墨烯分子和10个聚碳化二亚胺分子交联结构模型中交联反应形成的C-N键的键长在不同长度的分布概率图。
图6、实施例3中利用计算机模拟构建1个氧化石墨烯分子和10个聚碳化二亚胺分子交联结构模型中交联反应形成的C-N键的键长在不同长度的分布概率图。
图7、实施例4中利用计算机模拟构建10个氧化石墨烯分子和20个聚碳化二亚胺分子交联结构模型中交联反应形成的C-N键的键长在不同长度的分布概率图。
具体实施方式
下面通过实施例并结合附图对本发明进一步阐述,但本发明并不限于此。
本发明各实施例中所用的Materials Studio软件购自北京创腾科技有限公司,版本号为8.0SP1,包含Visualizer、Forcite、Amorphous Cell模块和COMPASS分子力场。
一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,构建过程逻辑框图如图1所示,即(1)利用MS软件的Visualizer模块分别构建氧化石墨烯分子、聚碳化二亚胺分子的结构,(2)利用MS软件的Forcite模块进行能量最小化运算,(3)利用MS软件的Amorphous Cell模块进行结构混合运算,(4)利用MS软件的Forcite模块进行能量最小化和分子动力学弛豫运算,(5)利用xLink程序进行虚拟交联反应运算,(6)利用MS软件中的Forcite模块进行温度循环的分子动力学弛豫运算;(7)根据步骤(6)所得交联结构的反应度与预先设定的反应度的目标值进行比较,判断反应度是否达到预先设定的反应度的目标值,当所得的交联结构的反应度达到预先设定的反应度的目标值,进行下一步,当所得的交联结构的反应度未达到预先设定的反应度的目标值,且当前交联反应距离小于 则将当前交联反应距离增大 然后重新进行执行步骤(5)-(7),直至交联结构D1的反应度达到预先设定的反应度的目标值;当所得的交联结构的反应度未达到预先设定的反应度的目标值,且当前交联反应距离达到交联反应距离最大值 重新进行步骤(3)-(7),直至交联结构D1的反应度达到预先设定的反应度的目标值;(8)测量并统计C-N键的键长,判断构建过程是否结束:如果C-N键的键长在 范围内,构建结束;如果C-N键的键长在 范围内,则重复步骤(6)-(8),直至反应度达到设定的目标值,且C-N键的键长为 如果C-N键的键长大于 则重复步骤(3)-(8),直至反应度达到预先设定的反应度的目标值,且C-N键的键长为 此时的交联结构即为氧化石墨烯与聚碳化二亚胺交联结构模型;
上述的利用xLink程序进行虚拟交联反应运算的逻辑框图如图2所示,即首先利用MS软件Visualizer模块的Measure工具测量结构中任意一对C-N原子对(所述的任意一对C-N原子对是指由氧化石墨烯分子中任一羰基基团中的C原子与聚碳化二亚胺分子中任一N=C=N基团中的N原子形成的原子对,后同)中C、N原子之间的距离D,判断此距离D是否小于或等于当前的交联反应距离,以此作为判断是否发生交联反应的依据;
当距离D小于或等于当前的交联反应距离时,发生交联反应,构建交联结构,即利用MS软件Visualizer模块将此距离D对应的羰基基团中的C原子与N=C=N基团中的N原子直接形成单键,将羰基基团中C原子上羟基基团中的O原子与N=C=N基团中的C原子直接形成双键,然后根据各原子配位情况进行H原子饱和;
当距离D大于当前的交联反应距离时,不发生交联反应,不进行操作;
对当前结构中所有C-N原子对均判断是否发生交联反应,直至当前结构中所有的C-N原子对均已完成判断,且所有符合发生交联反应条件的C-N原子对均已构建交联结构,交联反应运算结束。
一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,以2个氧化石墨烯分子和5个聚碳化二亚胺分子构建反应度为100%的交联结构为例,具体包括如下步骤:
(1)、利用MS软件中的Visualizer模块分别构建氧化石墨烯分子、聚碳化二亚胺分子的结构GO1、NCN1;
(2)、利用MS软件中的Forcite模块分别对步骤(1)所得的两个结构文件GO1、NCN1进行能量最小化运算,选用Smart Minimize方法、COMPASS分子力场、Atom based非键作用求和方法,分别得到能量最优的结构GO2、NCN2;
(3)、利用MS软件中的Amorphous Cell模块对步骤(2)所得的两个结构GO2、NCN2进行结构混合运算,其中分子力场选用COMPASS力场,库伦作用求和选用Ewald方法,范德华作用求和选用Atom based方法,氧化石墨烯分子加载数设为2个,聚碳化二亚胺分子加载数设为5个,总原子数为324个原子,得到混合结构Cell1;
(4)、利用MS软件中的Forcite模块对步骤(3)所得的混合结构Cell1进行能量最小化和分子动力学弛豫运算;
进行能量最小化运算时,选用Smart Minimize方法、COMPASS分子力场、Ewald非键作用求和方法;
进行分子动力学弛豫运算时,选用COMPASS分子力场、NPT系综、Ewald非键作用求和方法,温度选用200K,压力选用1大气压即1.0×10
(5)、利用xLink程序对步骤(4)运算所得结构进行“虚拟交联反应”运算;
交联反应距离初始值设定为 仅运行本步骤(5)时,此交联反应距离不做改变;经后续步骤(7)运算判断后,需要增大交联反应距离时,每次增大 交联反应距离的最大值设定为
利用MS软件Visualizer模块的Measure工具测量步骤(4)运算所得的结构中任意一对C-N原子对中C、N原子之间的距离D;
其中所述的C-N原子对是指由氧化石墨烯分子中羰基基团中的C原子与聚碳化二亚胺分子中N=C=N基团中的N原子形成的原子对;
当C、N原子之间的距离D小于或等于当前的交联反应距离时,发生交联反应,构建交联结构;
所述的构建交联结构,即利用MS软件Visualizer模块将上述发生交联反应的结构中对应的羰基基团中的C原子与N=C=N基团中的N原子直接形成单键,将羰基基团中的C原子上羟基基团中的O原子与N=C=N基团中的C原子直接形成双键,然后根据各原子配位情况进行H原子饱和;
当C、N原子之间的距离D大于当前的交联反应距离时,不发生交联反应,不进行操作;
再对另外一对C-N原子对运行本步骤(5),直至步骤(4)运算所得的结构中所有的C-N原子对均已判断是否发生交联反应,且所有符合上述发生交联反应条件的C-N原子对均已构建交联结构,得到交联结构D1;
(6)、利用MS软件中的Forcite模块对步骤(5)虚拟交联反应运算所得的交联结构D1进行温度循环的分子动力学弛豫运算得到交联结构D1的反应度;
交联结构D1的反应度定义为交联结构D1中所用的氧化石墨烯分子中已经发生交联反应的羰基基团的个数与交联结构D1中所用的氧化石墨烯分子中所有羰基基团个数的比值;
温度循环的分子动力学弛豫时,其中力场选用COMPASS分子力场,系综选用NVT系综,温度循环范围选用300K-500K-300K,温度变化量选用50K,每个温度下分子动力学运算的总模拟时间选用300ps;
即首先在温度300K条件下进行分子动力学弛豫运算,然后依次进行温度350、400、450、500K条件下的分子动力学弛豫运算,运算完成后再依次进行450、400、350、300K条件下的分子动力学弛豫运算;
每次分子动力学运算均以上一个分子动力学运算后的结果作为初始结构,每次分子动力学运算的总模拟时间相同,均为300ps,直至上述的温度由300K升温到500K后再降温到300K的分子动力学弛豫运算完成,结束温度循环的分子动力学弛豫运算,得到交联结构D1的反应度;
(7)、步骤(6)运算所得交联结构D1的反应度与预先设定的反应度的目标值(在步骤(5)的虚拟交联反应程序中设定)进行比较;
预先设定的反应度的目标值为100%,即所有氧化石墨烯中的羰基均发生交联反应;
当步骤(6)运算所得的交联结构D1的反应度达到100%,此时的交联结构即为满足要求的氧化石墨烯与聚碳化二亚胺交联结构;
当步骤(6)运算所得的交联结构D1的反应度未达到100%,且当前交联反应距离小于 则将当前交联反应距离增大 然后重新进行步骤(5)-(7),直至反应度达到100%;
当步骤(6)运算所得的交联结构D1的反应度未达到100%,且当前交联反应距离达到最大值 运算暂停,提示反应度未达到100%,重新进行步骤(3)-(7),直至反应度达到100%;
上述当交联结构D1的反应度达到100%时,所得交联结构D1中的部分交联结构示意图如图3所示,从图3中可以看出,氧化石墨烯中的羰基基团已与聚碳化二亚胺中的N=C=N基团通过“虚拟交联反应”形成酰基脲结构,由此表明本发明可以简单、快速的构建氧化石墨烯-聚碳化二亚胺交联结构;
(8)、步骤(7)中所得的交联结构D1的反应度达到预先设定的反应度的目标值后,利用MS软件Visualizer模块的Measure工具测量并统计所得的交联结构D1中的C-N键的键长L;
测量和统计的交联结构D1中的C-N键的键长分布结果如图4所示,从图4中可以看出,通过“虚拟交联反应”形成的C-N键的键长分布在 范围内,由此表明上述构建的交联结构合理,没有出现穿环等异常结构,构建结束。此时的交联结构D1即为以2个氧化石墨烯分子和5个聚碳化二亚胺分子构建的、反应度为100%的交联结构。
一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,以5个氧化石墨烯分子和10个聚碳化二亚胺分子构建反应度为80%的交联结构为例,具体包括如下步骤:
(1)、利用MS软件中的Visualizer模块分别构建氧化石墨烯分子、聚碳化二亚胺分子的结构GO1、NCN1;
(2)、利用MS软件中的Forcite模块分别对步骤(1)所得的两个结构文件GO1、NCN1进行能量最小化运算,选用Smart Minimize方法、COMPASS分子力场、Atom based非键作用求和方法,分别得到能量最优的结构GO2、NCN2;
(3)、利用MS软件中的Amorphous Cell模块对步骤(2)所得的两个结构GO2、NCN2进行结构混合运算,其中分子力场选用COMPASS力场,库伦作用求和选用Ewald方法,范德华作用求和选用Atom based方法,氧化石墨烯分子加载数设为5个,聚碳化二亚胺分子加载数设为10个,总原子数为735个原子,得到混合结构Cell1;
(4)、利用MS软件中的Forcite模块对步骤(3)所得的混合结构Cell1进行能量最小化和分子动力学弛豫运算;
进行能量最小化运算时,选用Smart Minimize方法、COMPASS分子力场、Ewald非键作用求和方法;
进行分子动力学弛豫运算时,选用COMPASS分子力场、NPT系综、Ewald非键作用求和方法,温度选用500K,压力选用1大气压即1.0×10
(5)、利用xLink程序对步骤(4)运算所得结构进行“虚拟交联反应”运算;
交联反应距离初始值设定为 仅运行本步骤(5)时,此交联反应距离不做改变;经后续步骤(7)运算判断后,需要增大交联反应距离时,每次增大 交联反应距离的最大值设定为
利用MS软件Visualizer模块的Measure工具测量步骤(4)运算所得的结构中任意一对C-N原子对中C、N原子之间的距离D;
其中所述的C-N原子对是指由氧化石墨烯分子中羰基基团中的C原子与聚碳化二亚胺分子中N=C=N基团中的N原子形成的原子对;
当C、N原子之间的距离D小于或等于当前的交联反应距离时,发生交联反应,构建交联结构;
所述的构建交联结构,即利用MS软件Visualizer模块将上述发生交联反应的结构中对应的羰基基团中的C原子与N=C=N基团中的N原子直接形成单键,将羰基基团中的C原子上羟基基团中的O原子与N=C=N基团中的C原子直接形成双键,然后根据各原子配位情况进行H原子饱和;
当C、N原子之间的距离D大于当前的交联反应距离时,不发生交联反应,不进行操作;
再对另外一对C-N原子对运行本步骤(5),直至步骤(4)运算所得的结构中所有的C-N原子对均已判断是否发生交联反应,且所有符合上述发生交联反应条件的C-N原子对均已构建交联结构,得到交联结构文件D1;
(6)、利用MS软件中的Forcite模块对步骤(5)虚拟交联反应运算所得的交联结构D1进行温度循环的分子动力学弛豫运算得到交联结构D1的反应度;
交联结构D1的反应度定义为交联结构D1中所用的氧化石墨烯分子中已经发生交联反应的羰基基团的个数与交联结构D1中所用的氧化石墨烯分子中所有羰基基团个数的比值;
温度循环的分子动力学弛豫时,其中力场选用COMPASS分子力场,系综选用NVT系综,温度循环范围选用300K-800K-300K,温度变化量选用100K,每个温度下分子动力学运算的总模拟时间选用1000ps;
即首先在温度300K条件下进行分子动力学弛豫运算,然后依次进行温度400、500、600、700、800K条件下的分子动力学弛豫运算,运算完成后再依次进行700、600、500、400、300K条件下的分子动力学弛豫运算;
每次分子动力学运算均以上一个分子动力学运算后的结果作为初始结构,每次分子动力学运算的总模拟时间相同,均为1000ps,直至上述的温度由300K升温到800K后再降温到300K的分子动力学弛豫运算完成,结束温度循环的分子动力学弛豫运算,得到交联结构D1的反应度;
(7)、步骤(6)运算所得交联结构D1的反应度与预先设定的反应度的目标值(在步骤(5)的虚拟交联反应程序中设定)进行比较;
预先设定的反应度的目标值为80%;
当步骤(6)运算所得的交联结构D1的反应度达到80%,此时的交联结构即为满足要求的氧化石墨烯与聚碳化二亚胺交联结构;
当步骤(6)运算所得的交联结构D1的反应度未达到80%,且当前交联反应距离小于 则将当前交联反应距离增大 然后重新进行步骤(5)-(7),直至交联结构D1的反应度达到80%;
当步骤(6)运算所得的交联结构D1的反应度未达到80%,且当前交联反应距离达到最大值 运算暂停,提示交联结构D1的反应度未达到80%,重新进行步骤(3)-(7),直至交联结构D1的反应度达到80%;
(8)、步骤(7)中所得的交联结构D1的反应度达到预先设定的反应度的目标值后,利用MS软件Visualizer模块的Measure工具测量并统计所得的交联结构D1中的C-N键的键长L;
测量和统计的交联结构D1中的C-N键的键长分布结果如图5所示,从图5中可以看出,通过“虚拟交联反应”形成的C-N键的键长分布在 范围内,由此表明上述步骤构建的交联结构合理,没有出现穿环等异常结构,构建结束,此时的交联结构D1即为以5个氧化石墨烯分子和10个聚碳化二亚胺分子构建的、反应度为80%的交联结构模型。
一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,以1个氧化石墨烯分子和10个聚碳化二亚胺分子构建反应度为100%的交联结构为例,具体包括如下步骤:
(1)、利用MS软件中的Visualizer模块分别构建氧化石墨烯分子、聚碳化二亚胺分子的结构GO1、NCN1;
(2)、利用MS软件中的Forcite模块分别对步骤(1)所得的两个结构文件GO1、NCN1进行能量最小化运算,选用Smart Minimize方法、COMPASS分子力场、Atom based非键作用求和方法,分别得到能量最优的结构GO2、NCN2;
(3)、利用MS软件中的Amorphous Cell模块对步骤(2)所得的两个结构GO2、NCN2进行结构混合运算,其中分子力场选用COMPASS力场,库伦作用求和选用Ewald方法,范德华作用求和选用Atom based方法,氧化石墨烯分子加载数设为1个,聚碳化二亚胺分子加载数设为10个,总原子数为387个原子,得到混合结构Cell1;
(4)、利用MS软件中的Forcite模块对步骤(3)所得的混合结构Cell1进行能量最小化和分子动力学弛豫运算;
进行能量最小化运算时,选用Smart Minimize方法、COMPASS分子力场、Ewald非键作用求和方法;
进行分子动力学弛豫运算时,选用COMPASS分子力场、NPT系综、Ewald非键作用求和方法,温度选用300K,压力选用1大气压即1.0×10
(5)、利用xLink程序对步骤(4)运算所得结构进行“虚拟交联反应”运算;
交联反应距离初始值设定为 仅运行本步骤(5)时,此交联反应距离不做改变;经后续步骤(7)运算判断后,需要增大交联反应距离时,每次增大 交联反应距离的最大值设定为
利用MS软件Visualizer模块的Measure工具测量步骤(4)运算所得的结构中任意一对C-N原子对中C、N原子之间的距离D;
其中所述的C-N原子对是指由氧化石墨烯分子中羰基基团中的C原子与聚碳化二亚胺分子中N=C=N基团中的N原子形成的原子对;
当C、N原子之间的距离D小于或等于当前的交联反应距离时,发生交联反应,构建交联结构;
所述的构建交联结构,即利用MS软件Visualizer模块将上述发生交联反应的结构中对应的羰基基团中的C原子与N=C=N基团中的N原子直接形成单键,将羰基基团中的C原子上羟基基团中的O原子与N=C=N基团中的C原子直接形成双键,然后根据各原子配位情况进行H原子饱和;
当C、N原子之间的距离D大于当前的交联反应距离时,不发生交联反应,不进行操作;
再对另外一对C-N原子对运行本步骤(5),直至步骤(4)运算所得的结构中所有的C-N原子对均已判断是否发生交联反应,且所有符合上述发生交联反应条件的C-N原子对均已构建交联结构,得到交联结构D1;
(6)、利用MS软件中的Forcite模块对步骤(5)虚拟交联反应运算所得的交联结构D1进行温度循环的分子动力学弛豫运算得到交联结构D1的反应度;
交联结构D1的反应度定义为交联结构D1中所用的氧化石墨烯分子中已经发生交联反应的羰基基团的个数与交联结构D1中所用的氧化石墨烯分子中所有羰基基团个数的比值;
温度循环的分子动力学弛豫时,其中力场选用COMPASS分子力场,系综选用NVT系综,温度循环范围选用300K-600K-300K,温度变化量选用75K,每个温度下分子动力学运算的总模拟时间选用500ps;
即首先在温度300K条件下进行分子动力学弛豫运算,然后依次进行温度375、450、525、600K条件下的分子动力学弛豫运算,运算完成后再依次进行525、450、375、300K条件下的分子动力学弛豫运算;
每次分子动力学运算均以上一个分子动力学运算后的结果作为初始结构,每次分子动力学运算的总模拟时间相同,均为500ps,直至上述的温度由300K升温到600K后再降温到300K的分子动力学弛豫运算完成,结束温度循环的分子动力学弛豫运算,得到交联结构D1的反应度;
(7)、步骤(6)运算所得交联结构D1的反应度与预先设定的反应度的目标值(在步骤(5)的虚拟交联反应程序中设定)进行比较;
预先设定的反应度的目标值为100%,即所有氧化石墨烯中的羰基均发生交联反应;
当步骤(6)运算所得的交联结构D1的反应度达到100%,此时的交联结构即为满足要求的氧化石墨烯与聚碳化二亚胺交联结构;
当步骤(6)运算所得的交联结构D1的反应度未达到100%,且当前交联反应距离小于 则将当前交联反应距离增大 然后重新进行步骤(5)-(7),直至反应度达到100%;
当步骤(6)运算所得的交联结构D1的反应度未达到100%,且当前交联反应距离达到最大值 运算暂停,提示反应度未达到100%,重新进行步骤(3)-(7),直至反应度达到100%;
(8)、步骤(7)中所得的交联结构D1的反应度达到预先设定的反应度的目标值后,利用MS软件Visualizer模块的Measure工具测量并统计所得的交联结构D1中的C-N键的键长L;
测量和统计的交联结构D1中的C-N键的键长分布结果如图6所示,从图6中可以看出,通过“虚拟交联反应”形成的C-N键的键长分布在 范围内,由此表明上述步骤构建的交联结构合理,没有出现穿环等异常结构,构建结束。此时的交联结构D1即为以1个氧化石墨烯分子和10个聚碳化二亚胺分子构建的、反应度为100%的交联结构模型。
一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,以10个氧化石墨烯分子和20个聚碳化二亚胺分子构建反应度为100%的交联结构为例,具体包括如下步骤:
(1)、利用MS软件中的Visualizer模块分别构建氧化石墨烯分子、聚碳化二亚胺分子的结构GO1、NCN1;
(2)、利用MS软件中的Forcite模块分别对步骤(1)所得的两个结构文件GO1、NCN1进行能量最小化运算,选用Smart Minimize方法、COMPASS分子力场、Atom based非键作用求和方法,分别得到能量最优的结构GO2、NCN2;
(3)、利用MS软件中的Amorphous Cell模块对步骤(2)所得的两个结构GO2、NCN2进行结构混合运算,其中分子力场选用COMPASS力场,库伦作用求和选用Ewald方法,范德华作用求和选用Atom based方法,氧化石墨烯分子加载数设为10个,聚碳化二亚胺分子加载数设为20个,总原子数为1470个原子,得到混合结构Cell1;
(4)、利用MS软件中的Forcite模块对步骤(3)所得的混合结构Cell1进行能量最小化和分子动力学弛豫运算;
进行能量最小化运算时,选用Smart Minimize方法、COMPASS分子力场、Ewald非键作用求和方法;
进行分子动力学弛豫运算时,选用COMPASS分子力场、NPT系综、Ewald非键作用求和方法,温度选用400K,压力选用1大气压即1.0×10
(5)、利用xLink程序对步骤(4)运算所得结构进行“虚拟交联反应”运算;
交联反应距离初始值设定为 仅运行本步骤(5)时,此交联反应距离不做改变;经后续步骤(7)运算判断后,需要增大交联反应距离时,每次增大 交联反应距离的最大值设定为
利用MS软件Visualizer模块的Measure工具测量步骤(4)运算所得的结构中任意一对C-N原子对中C、N原子之间的距离D;
其中所述的C-N原子对是指由氧化石墨烯分子中羰基基团中的C原子与聚碳化二亚胺分子中N=C=N基团中的N原子形成的原子对;
当C、N原子之间的距离D小于或等于当前的交联反应距离时,发生交联反应,构建交联结构;
所述的构建交联结构,即利用MS软件Visualizer模块将上述发生交联反应的结构中对应的羰基基团中的C原子与N=C=N基团中的N原子直接形成单键,将羰基基团中的C原子上羟基基团中的O原子与N=C=N基团中的C原子直接形成双键,然后根据各原子配位情况进行H原子饱和;
当C、N原子之间的距离D大于当前的交联反应距离时,不发生交联反应,不进行操作;
再对另外一对C-N原子对运行本步骤(5),直至步骤(4)运算所得的结构中所有的C-N原子对均已判断是否发生交联反应,且所有符合上述发生交联反应条件的C-N原子对均已构建交联结构,得到交联结构D1;
(6)、利用MS软件中的Forcite模块对步骤(5)虚拟交联反应运算所得的交联结构D1进行温度循环的分子动力学弛豫运算得到交联结构D1的反应度;
交联结构D1的反应度定义为交联结构D1中所用的氧化石墨烯分子中已经发生交联反应的羰基基团的个数与交联结构D1中所用的氧化石墨烯分子中所有羰基基团个数的比值;
温度循环的分子动力学弛豫时,其中力场选用COMPASS分子力场,系综选用NVT系综,温度循环范围选用300K-620K-300K,温度变化量选用80K,每个温度下分子动力学运算的总模拟时间选用800ps;
即首先在温度300K条件下进行分子动力学弛豫运算,然后依次进行温度380、460、540、620K条件下的分子动力学弛豫运算,运算完成后再依次进行540、460、380、300K条件下的分子动力学弛豫运算;
每次分子动力学运算均以上一个分子动力学运算后的结果作为初始结构,每次分子动力学运算的总模拟时间相同,均为800ps,直至上述的温度由300K升温到620K后再降温到300K的分子动力学弛豫运算完成,结束温度循环的分子动力学弛豫运算,得到交联结构D1的反应度;
(7)、步骤(6)运算所得交联结构D1的反应度与预先设定的反应度的目标值(在步骤(5)的虚拟交联反应程序中设定)进行比较;
预先设定的反应度的目标值为100%,即所有氧化石墨烯中的羰基均发生交联反应;
当步骤(6)运算所得的交联结构D1的反应度达到100%,此时的交联结构即为满足要求的氧化石墨烯与聚碳化二亚胺交联结构;
当步骤(6)运算所得的交联结构D1的反应度未达到100%,且当前交联反应距离小于 则将当前交联反应距离增大 然后重新进行步骤(5)-(7),直至反应度达到100%;
当步骤(6)运算所得的交联结构D1的反应度未达到100%,且当前交联反应距离达到最大值 运算暂停,提示反应度未达到100%,重新进行步骤(3)-(7),直至反应度达到100%;
(8)、步骤(7)中所得的交联结构D1的反应度达到预先设定的反应度的目标值后,利用MS软件Visualizer模块的Measure工具测量并统计所得的交联结构D1中的C-N键的键长L;
测量和统计的交联结构D1中的C-N键的键长分布结果如图7所示,从图7中可以看出,通过“虚拟交联反应”形成的C-N键的键长分布在 范围内,由此表明上述步骤构建的交联结构合理,没有出现穿环等异常结构,构建结束。此时的交联结构D1即为以10个氧化石墨烯分子和20个聚碳化二亚胺分子构建的、反应度为100%的交联结构模型。
综上所述,本发明提供的一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法,通过分子结构构建、能量最小化、结构混合运算、能量最小化和200-500K温度条件的分子动力学弛豫运算、“虚拟交联反应”运算、300-(500-800)-300K温度循环的分子动力学弛豫运算,构建了指定反应度的氧化石墨烯-聚碳化二亚胺交联结构模型,并且通过键长统计判断该交联结构模型并未产生穿环等不合理结构。
以上所述仅是本发明的实施方式的举例,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明
一种利用计算机模拟构建氧化石墨烯与聚碳化二亚胺交联结构模型的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0