

IPC分类号 : C07D333/72,C07D345/00,A61K31/381,A61K31/33,A61P7/02,A61P9/10,A61P9/00,A61P3/06,A61P3/10,A61P3/00,A61P35/00








专利摘要
本发明涉及药物化学和药物治疗学领域,具体涉及一种3-取代苯并[c]呋喃酮的硫代、硒代同系物(I)。该类化合物可用于制备预防和治疗心脑缺血性疾病及改善心脑循环障碍药物、抗血小板药物、抗血栓药物、抗脑缺血药物、降血脂药物、抗动脉粥样硬化药物、抗糖尿病及其并发症药物、治疗代谢综合症药物或抗肿瘤药物。本发明还涉及这类化合物的制备方法以及含有它们的药物组合。
说明书
技术领域
本发明涉及药物化学和药物治疗学领域,具体涉及一种3-取代苯并[c]呋喃酮的硫代、硒代同系物。该类化合物可用于制备预防和治疗心脑缺血性疾病及改善心脑循环障碍药物、抗血小板药物、抗血栓药物、抗脑缺血药物、降血脂药物、抗动脉粥样硬化药物、抗糖尿病及其并发症药物、治疗代谢综合症药物或抗肿瘤药物。本发明还涉及这类化合物的制备方法以及含有它们的药物组合。
背景技术
血小板活性异常在心脑血管疾病的发生及发展中起着非常重要的作用。在正常血液循环中血小板处于静息状态。当血管内皮损伤或在某些生理病理刺激因子作用下,血小板发生粘附、聚集等活化反应。
高血压病患者血小板表面的血管紧张素II(Ang II)受体具有强烈的刺激血小板聚集和释放作用,可导致血小板活化、血管内皮细胞受损和血栓性并发症(Hypertension,1993,22,853)。
高血脂和动脉粥样硬化患者的血液高粘稠度以及低密度脂蛋白的氧化可活化血小板,继而在流经动脉粥样块斑处时粘附聚集,释放出一系列的活性致聚物质,进而在纤维蛋白原及Ca2+的作用下,发生聚集反应,从而形成血栓(Trends Cardiovasc.Med.,2004,14,18)。
对于充血性心力衰竭病人,缺氧引起的血管内皮细胞损伤及多种细胞因子释放均能导致血小板活性异常,使得发生血栓性并发症的几率增加(N.Engl.J.Med.2001,345,1689)。
缺血性脑血管疾病患者的血小板活化程度明显增加,引发血小板在脑部动脉血管中聚集,导致血栓形成,动脉闭塞,最后造成脑组织器官坏死。血小板活化程度与梗死灶体积呈正相关(Blood,2008,112,3555)。
此外,患有冠心病、心律失常、炎症性疾病、糖尿病等病人的血小板功能异常均具有普遍性和贯穿整个病程的特点。因此,抗血小板聚集治疗成为心脑血管疾病治疗的重要手段。
苯并[c]呋喃酮类化合物,又称为苯酞类化合物,在植物界资源丰富,是许多常用中草药(如当归、川芎、芹菜等)的主要有效成分,其具有抗炎、抗菌、抗肿瘤、抗病毒、抗氧化及心血管保护等多种生物活性(J.Nat.Prod.,2007,70,891)。其中,3-正丁基苯并[c]呋喃酮(又称为丁苯酞)与芹菜籽中提取的左旋芹菜甲素的结构相同,为其人工合成的消旋体,在我国于2004年11月批准上市。临床研究结果表明,丁苯酞与丹参注射液静脉滴注联合应用对急性缺血性脑卒中患者的中枢神经功能的损伤有改善作用,可促进患者功能恢复。动物药效学研究提示,丁苯酞可阻断缺血性脑卒中所致脑损伤的多个病理环节,具有较强的抗脑缺血和脑保护作用,尤其是能够明显提高缺血小鼠脑内ATP和磷酸肌酸水平,明显缩小大鼠局部脑缺血的梗塞面积,减轻脑水肿,改善脑能量代谢和缺血脑区的微循环和血流量,抑制神经细胞凋亡,并具有一定的抗脑血栓形成和抗血小板聚集作用(J.Neurol.Sci.,2007,260,106)。另有文献表明,丁基苯酞通过影响花生四烯酸(AA)代谢,选择性抑制AA及其代谢产物介导的多种病理生理过程,可以解除微血管痉挛,抑制血小板聚集,抑制血栓烷A2的合成,清除自由基等,从而通过多途径,多环节阻断脑缺血引起的病理生理发展过程,保护神经元,修复神经功能。(J.Cardiovasc.Pharmacol.,2004,43,876;Acta Pharmacol.Sin.,1998,19,117)。
丁苯酞
然而,丁苯酞抗血栓作用弱于阿司匹林,临床上常将其与某种抗血栓药物联合使用(心脑血管病防治,2009,9,29)。因此,为了增强丁苯酞的抗血栓效果,需要研究和开发具有新的结构特征和新的作用机理的新型化合物。
发明内容
本发明公开了一种3-取代苯并[c]呋喃酮的硫代、硒代同系物或其对映体、非对映异构体及其药学上可接受的盐,药理实验证明,该类化合物具有良好的抑制血小板聚集活性,因此,它们可用于预防和治疗与血小板活性异常的相关疾病,这类疾病包括缺血性脑血管疾病、高血脂症、动脉粥样硬化、高血压、充血性心率衰竭、冠心病、糖尿病及其并发症、代谢综合症等。
本发明化合物结构如通式I所示,其具有对映体和非对映异构体结构。
其中:
X代表硫原子时,R代表C4-C10直链烷基、C3-C10支链烷基或(CH2)n苯基,其中n代表0-6的整数;
X代表硒原子时,R代表C1-C10直链烷基、C3-C10支链烷基或(CH2)n苯基,其中n代表0-6的整数。
具体的说,通式I所述化合物或其对映体、非对映异构体及其药学上可接受的盐,其特征在于所述通式I中:
X代表硫原子或硒原子;
R代表C4-C8直链烷基、C3-C8支链烷基或苄基。
进-步地,通式I所述化合物优选自下列化合物:
3-正丁基苯并[c]噻吩酮;
3-正戊基苯并[c]噻吩酮;
3-异戊基苯并[c]噻吩酮;
3-正己基苯并[c]噻吩酮;
3-正庚基苯并[c]噻吩酮;
3-正辛基苯并[c]噻吩酮;
3-苄基苯并[c]噻吩酮;
3-乙基苯并[c]硒吩酮;
3-正丙基苯并[c]硒吩酮;
3-正丁基苯并[c]硒吩酮;
3-正戊基苯并[c]硒吩酮;
3-异戊基苯并[c]硒吩酮;
3-正己基苯并[c]硒吩酮;
3-正庚基苯并[c]硒吩酮;
3-正辛基苯并[c]硒吩酮;
3-苄基苯并[c]硒吩酮。
具体地讲,通式I所述化合物进一步优选自下列化合物:
3-正丁基苯并[c]噻吩酮(化合物编号:I1,下同);
3-正戊基苯并[c]噻吩酮(I2);
3-异戊基苯并[c]噻吩酮(I3);
3-正己基苯并[c]噻吩酮(I4);
3-正庚基苯并[c]噻吩酮(I5);
3-正丙基苯并[c]硒吩酮(I6);
3-正丁基苯并[c]硒吩酮(I7);
3-正戊基苯并[c]硒吩酮(I8);
3-异戊基苯并[c]硒吩酮(I9);
3-正己基苯并[c]硒吩酮(I10);
3-正庚基苯并[c]硒吩酮(I11)。
本发明优选化合物的对映体、非对映异构体及其药学上可接受的盐构成了本发明的完整部分。
本发明的另一目的在于提供本发明通式I所述化合物的制备方法,其特征为在于,包括如下步骤:
a)当X为硫原子时,通式I所示的3-取代苯并[c]呋喃酮的硫代同系物的制备方法包括如下步骤:
以邻苯二甲酸酐为起始原料,与九水合硫化钠混合搅拌5h,加水和稀盐酸得到苯并[c]噻吩-1,3-二酮(II),II与相应的格氏试剂(RMgBr)反应制得化合物(III),III最后在氢碘酸作用下制得目标化合物(I);合成路线如下:
其中,R的定义如前所述;
b)当X为硒原子时,通式I所示的3-取代苯并[c]呋喃酮的硒代同系物的制备包括如下步骤:
以邻苯二甲酰氯为起始原料,将其加入硒粉和氢化铝锂的混合液中搅拌5h,加水和稀盐酸得到苯并[c]硒吩-1,3-二酮(IV);IV与相应的格氏试剂(RMgBr)反应得化合物(V),V最后在氢碘酸作用下转化为目标物(I);合成路线如下:
其中,R的定义如前所述;
制备式(V)化合物的特征在于,格氏试剂(RMgBr)与苯并[c]硒吩-1,3-二酮(IV)投料量的摩尔比为2-3;采用的反应温度为25-70℃;制备目标化合物(I)的特征在于,氢碘酸与化合物(III)或(V)投料量的摩尔比为3-5;采用的反应温度为100-125℃。
这些化合物可按照常规分离技术加以纯化,如果需要可用常规分离技术分离成它们的异构体。
本发明的进一步目的在于提供一种含有效剂量的通式I化合物或其对映体、非对映异构体及其药学上可接受的盐及载体的药物组合物。
本发明的再一目的是提供本发明通式I化合物在制备预防或治疗与血小板聚集有关疾病药物中的应用,尤其是用于制备预防和治疗心脑缺血性疾病及改善心脑循环障碍药物、抗血小板药物、抗血栓药物、抗脑缺血药物、降血脂药物、抗动脉粥样硬化药物、抗糖尿病及其并发症药物、治疗代谢综合症药物或抗肿瘤药物。
与现有技术相比,本发明有益效果在于:硫和硒与氧为同族元素,选择性地将化合物分子中的某一氧原子用硫或硒原子取代,通常会改善分子的亲脂性,并引起分子中静电性的变化;含有硫或硒原子的药物与一般药物相比,具有更好的细胞膜穿透性和靶蛋白的特定选择性;此外,硫或硒原子的引入还可能使分子具有抗氧化作用和神经保护作用。
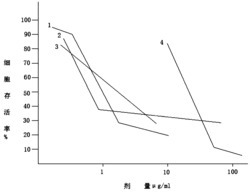
本发明化合物对血小板聚集活性的药理实验方法与结果如下:
实验方法:取家兔2只,用利多卡因局部麻醉,手术分离颈总动脉取血,采取3.8%枸橼酸钠1∶9抗凝,以500r/min离心10min,制备富血小板血浆(PRP),剩余部分再以3000r/min离心,制备贫血小板血浆(PPP),按比浊法进行血小板聚集实验。测定管中加入PRP 240μL和不同浓度受试药物30μL,温孵5min,分别以30μL二磷酸腺苷钠盐(ADP)(终浓度为10μmol/L)和30μL花生四烯酸(AA)(终浓度为1mmol/L)为诱导剂,观察记录5min内最大聚集率。用生理盐水(NS)作对照,计算各受试化合物的抑制率(%):
抑制率(%)=[对照组5min内最大聚集率-待测样品组5min内最大聚集率]/[对照组5min内最大聚集率]×100%。
测试结果:表1中列出了部分化合物对ADP和AA诱导的家兔血小板聚集活性数据,阳性对照药为正丁基苯酞(NBP)。
表1.本发明部分化合物对ADP和AA诱导的家兔血小板聚集的抑制活性(n=4, )
*P<0.05,**P<0.01 vs Control group;#P<0.05,##P<0.01,###P<0.001 vs NBP group.
α受试化合物浓度为100μM;
以上药理学数据显示,本发明涉及的3-取代苯并[c]呋喃酮的硫、硒代同系物能够不同程度的抑制血小板聚集,其中,化合物I1、I2、I4、I5、I10和I11与阳性对照药NBP相比,能更好的抑制血小板聚集。
具体实施方式
为了进一步阐明本发明,下面给出一系列实施例,这些实施例完全是例证性的,它们仅用来对本发明具体描述,不应当理解为对本发明的限制。
实施例1
苯并[c]噻吩-1,3-二酮的合成
在干燥的圆底烧瓶中加入15.0g(0.1mol)邻苯二甲酸酐和30.0g(0.125mol)九水合硫化钠,室温下机械搅拌5h,整个过程中有大量硫化氢气体产生且反应体系逐渐变成液体。反应完毕缓慢加入200mL蒸馏水,室温下继续搅拌0.5小时。向该反应体系中加入100mL 5%稀盐酸,有大量白色固体析出,静置过夜。过滤收集析出的固体,水洗、干燥,得到白色粉末状标题化合物14.9g,收率92%,m.p.112-114℃。ESI-MS:m/z 187[M+Na]+;1H NMR(300MHz,CDCl3,δ):7.74-7.79(m,2H,ArH),7.92-7.95(m,2H,ArH).
3-羟基-3-乙基苯并[c]噻吩酮的合成
在干燥的三颈瓶中放入4.84g(0.20mol)镁粉和20mL无水乙醚,加入少量溴乙烷以引发反应,然后继续滴加溴乙烷直至镁粉完全消失,溴乙烷的加入总量为21.8g(0.20mol),回流1h,冷却待用。向250mL含16.4g(0.10mol)苯并[c]噻吩-1,3-二酮的无水乙醚溶液中逐滴加入该格氏试剂,滴毕,加热回流1h,冷却室温搅拌8h。向此混合物中小心加入150mL饱和氯化铵溶液淬灭反应,然后加入5%稀盐酸调pH 2-3,分出有机相,水相再用乙醚萃取三次,合并有机相,无水硫酸钠干燥后蒸去溶剂。快速柱层析(石油醚/乙酸乙酯:10/1,V/V)得到黄色粉末状标题化合物10.8g,收率56%,m.p.92-94℃。ESI-MS:m/z 193[M-H]-;1H NMR(CDCl3,300Hz,δ):1.03(t,3H,CH3,J=7.1Hz),2.07-2.12(m,1H,CH),2.31-2.39(m,1H,CH),3.53(brs,1H,OH),7.47(m,1H,ArH),7.59-7.64(m,2H,ArH),7.83(d,1H,ArH,J=7.5Hz).
3-乙基苯并[c]噻吩酮的合成
将5.0g(25.7mmol)3-羟基-3-乙基苯并[c]噻吩酮溶于10mL冰醋酸中,加入5mL 57%氢碘酸溶液,升温至125℃搅拌0.5-1h。冷却后向反应混合液中加入150mL 5%亚硫酸氢钠溶液,水层用乙醚萃取三次,合并有机相,无水硫酸钠干燥后蒸去溶剂。快速柱层析(石油醚/乙酸乙酯:50/1,V/V)得到淡黄色油状标题化合物2.11g,收率46%。ESI-MS m/z:201[M+Na]+;1H NMR(CDCl3,300Hz,δ):1.05(t,3H,CH3,J=7.2Hz),1.84-1.95(m,1H,CH),2.30-2.38(m,1H,CH),4.83(m,1H,SCH),7.45(m,1H,ArH),7.52(d,1H,ArH,J=7.5Hz),7.64(m,1H,ArH),7.80(d,1H,ArH,J=7.5Hz).
实施例2
3-羟基-3-正丙基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用3.50g(21.3mmol)苯并[c]噻吩-1,3-二酮、5.31g(42.7mmol)溴代正丙烷和1.09g(42.7mmol)镁粉进行反应,得到黄色粉末状标题化合物2.26g,收率51%,m.p.78-80℃。ESI-MS:m/z 207[M-H]-;1H NMR(CDCl3,300Hz,δ):1.03(t,3H,CH3,J=7.1Hz),1.45-1.57(m,2H,CH2),2.03-2.10(m,1H,CH),2.22-2.33(m,1H,CH),3.51-3.60(br s,1H,OH),7.42(m,1H,ArH),7.52-7.67(m,2H,ArH),7.80(d,1H,ArH,J=7.5Hz).
3-正丙基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用2.51g(12.0mmol)3-羟基-3-正丙基苯并[c]噻吩酮与2mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物1.29g,收率56%。ESI-MS m/z:193[M+H]+;1H NMR(CDCl3,300Hz,δ):0.99(t,3H,CH3,J=7.5Hz),1.43-1.61(m,2H,CH2),1.73-1.84(m,1H,CH),2.20-2.30(m,1H,CH),4.84(m,1H,SCH),7.45(m,1H,ArH),7.52(d,1H,ArH,J=7.5Hz),7.65(m,1H,ArH),7.79(d,1H,ArH,J=7.5Hz).
实施例3
3-羟基-3-正丁基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用6.55g(39.9mmol)苯并[c]噻吩-1,3-二酮、10.9g(79.9mmol)溴代正丁烷和1.92g(79.9mmol)镁粉进行反应,得到黄色粉末状标题化合物5.41g,收率61%,m.p.65-67℃。ESI-MS:m/z 221[M-H]-;1H NMR(CDCl3,300Hz,δ):0.91(t,3H,CH3,J=7.1Hz),1.12-1.73(m,4H,2×CH2),2.07-2.17(m,1H,CH),2.35-2.45(m,1H,CH),3.58(br s,1H,OH),7.48(m,1H,ArH),7.62-7.68(m,2H,ArH),7.80(d,1H,ArH,J=7.8Hz).
3-正丁基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用4.20g(18.9mmol)3-羟基-3-正丁基苯并[c]噻吩酮与4mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物2.64g,收率67%。ESI-MS m/z:207[M+H]+;1H NMR(CDCl3,300Hz,δ):0.92(t,3H,CH3,J=7.2Hz),1.35-1.56(m,4H,2×CH2),1.74-1.87(m,1H,CH),2.26-2.35(m,1H,CH),4.83(m,1H,SCH),7.45(m,1H,ArH),7.53(d,1H,ArH,J=7.5Hz),7.65(m,1H,ArH),7.79(d,1H,ArH,J=7.5Hz).
实施例4
3-羟基-3-正戊基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用5.54g(33.8mmol)苯并[c]噻吩-1,3-二酮、10.2g(67.6mmol)溴代正戊烷和1.62g(67.6mmol)镁粉进行反应,得到黄色粉末状标题化合物5.10g,收率64%,m.p.61-63℃。ESI-MS:m/z 235[M-H]-;1H NMR(CDCl3,300Hz,δ):0.90(t,3H,CH3,J=6.9Hz),1.22-1.67(m,6H,3×CH2),2.06-2.15(m,1H,CH),2.34-2.45(m,1H,CH),3.60(br s,1H,OH),7.45(m,1H,ArH),7.52-7.68(m,2H,ArH),7.80(d,1H,ArH,J=7.8Hz).
3-正戊基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用3.24g(13.7mmol)3-羟基-3-正戊基苯并[c]噻吩酮与3mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物1.96g,收率65%。ESI-MS:m/z 221[M+H]+;243[M+Na]+;1H NMR(CDCl3,300Hz,δ):0.89(t,3H,CH3,J=6.9Hz),1.32-1.57(m,6H,3×CH2),1.75-1.85(m,1H,CH),2.23-2.33(m,1H,CH),4.83(m,1H,SCH),7.45(m,1H,ArH),7.52(d,1H,ArH,J=7.5Hz),7.62(m,1H,ArH),7.79(d,1H,ArH,J=7.5Hz).
实施例5
3-羟基-3-异戊基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用4.52g(27.6mmol)苯并[c]噻吩-1,3-二酮、9.09g(55.1mmol)溴代异戊烷和1.32g(55.1mmol)镁粉进行反应,得到黄色油状标题化合物3.97g,收率61%。ESI-MS:m/z 235[M-H]-;1H NMR(CDCl3,300Hz,δ):0.90(m,6H,2×CH3),1.23-1.52(m,3H,CH2,CH),2.07-2.12(m,1H,CH),2.34-2.44(m,1H,CH),3.55(br s,1H,OH),7.47(m,1H,ArH),7.53-7.63(m,2H,ArH),7.78(d,1H,ArH,J=7.8Hz).
3-异戊基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用3.04g(12.9mmol)3-羟基-3-异戊基苯并[c]噻吩酮与3mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物1.95g,收率69%。ESI-MS:m/z 221[M+H]+;1H NMR(CDCl3,300Hz,δ):0.90(m,6H,2×CH3),1.22-1.30(m,2H,CH2),1.48(m,1H,CH),1.74-1.87(m,1H,CH),2.24-2.35(m,1H,CH),4.83(m,1H,SCH),7.45(m,1H,ArH),7.53(d,1H,ArH,J=7.5Hz),7.63(m,1H,ArH),7.79(d,1H,ArH,J=7.5Hz).
实施例6
3-羟基-3-正己基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用7.34g(44.7mmol)苯并[c]噻吩-1,3-二酮、14.7g(89.5mmol)溴代正己烷和2.15g(89.5mmol)镁粉进行反应,得到黄色粉末状标题化合物7.27g,收率65%,m.p.56-58℃。ESI-MS:m/z 249[M-H]-;1H NMR(CDCl3,300Hz,δ):0.89(t,3H,CH3,J=7.1Hz),1.32-1.57(m,8H,4×CH2),2.00-2.04(m,1H,CH),2.30-2.42(m,1H,CH),3.58(br s,1H,OH),7.46(m,1H,ArH),7.52-7.63(m,2H,ArH),7.80(d,1H,ArH,J=7.8Hz).
3-正己基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用6.45g(25.8mmol)3-羟基-3-正己基苯并[c]噻吩酮与6mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物3.56g,收率59%。ESI-MS:m/z 235[M+H]+;257[M+Na]+;1H NMR(CDCl3,300Hz,δ):0.90(t,3H,CH3,J=7.2Hz),1.30-1.58(m,8H,4×CH2),1.74-1.86(m,1H,CH),2.23-2.34(m,1H,CH),4.83(m,1H,SCH),7.45(m,1H,ArH),7.52(d,1H,ArH,J=7.5Hz),7.62(m,1H,ArH),7.79(d,1H,ArH,J=7.5Hz).
实施例7
3-羟基-3-正庚基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用6.50g(39.6mmol)苯并[c]噻吩-1,3-二酮、14.2g(79.3mmol)溴代正庚烷和1.90g(79.3mmol)镁粉进行反应,得到黄色油状标题化合物4.70g,收率45%,m.p.65-67℃。ESI-MS:m/z 263[M-H]-;1H NMR(CDCl3,300Hz,δ):0.90(t,3H,CH3,J=7.2Hz),1.31-1.60(m,10H,5×CH2),2.02-2.05(m,1H,CH),2.30-2.44(m,1H,CH),3.56(br s,1H,OH),7.45(m,1H,ArH),7.52-7.62(m,2H,ArH),7.77(d,1H,ArH,J=7.5Hz).
3-正庚基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用4.45g(16.8mmol)3-羟基-3-正己基苯并[c]噻吩酮与4mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物2.05g,收率49%。ESI-MS:m/z 249[M+H]+;1H NMR(CDCl3,300Hz,δ):0.85(t,3H,CH3,J=7.2Hz),1.27-1.58(m,10H,5×CH2),1.74-1.86(m,1H,CH),2.23-2.34(m,1H,CH),4.83(m,1H,SCH),7.45(m,1H,ArH),7.52(d,1H,ArH,J=7.5Hz),7.62(m,1H,ArH),7.79(d,1H,ArH,J=7.5Hz).
实施例8
3-羟基-3-苄基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用3.53g(21.5mmol)苯并[c]噻吩-1,3-二酮、5.44g(43,0mmol)氯苄和1.03g(43.0mmol)镁粉进行反应,得到黄色粉末状标题化合物4.24g,收率77%,m.p.126-128℃。ESI-MS:m/z 255[M-H]-;1H NMR(CDCl3,300Hz,δ):3.48-3.69(m,2H,CH2),7.26-7.36(m,5H,ArH),7.48(m,1H,ArH),7.62(m,2H,ArH),7.68(d,1H,ArH,J=8.1Hz).
3-苄基苯并[c]噻吩酮的合成
按实施例1的类似方法,使用4.05g(15.8mmol)3-羟基-3-苄基苯并[c]噻吩酮与4mL 57%氢碘酸溶液反应,得到淡黄色粉末状标题化合物2.35g,收率62%,m.p.118-120℃。ESI-MS:m/z 241[M+H]+;1H NMR(CDCl3,300Hz,δ):3.02-3.09(m,1H,CH),3.53-3.59(m,1H,CH),5.07(m,1H,SCH),7.24-7.36(m,5H,ArH),7.47(m,2H,ArH),7.62(m,1H,ArH),7.80(d,1H,ArH,J=8.1Hz).
实施例9
苯并[c]硒吩-1,3-二酮的合成
在含无水四氢呋喃50mL的250mL圆底烧瓶中加入硒粉10.4g(0.13mol)和氢化铝锂5.0g(0.13mol),0℃下搅拌0.5h。向反应体系中加入邻苯二甲酰氯26.4g(0.13mol),室温搅拌5h。反应结束小心加入150mL水,用乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥后蒸去溶剂。快速柱层析(石油醚/乙酸乙酯:20/1,V/V),得到黄色粉末状标题化合物14.5g,收率53%,m.p.125-126℃。ESI-MS:m/z 235[M+Na]+;1H NMR(300MHz,CDCl3,δ):7.75-7.79(m,2H,ArH),795-7.99(m,2H,ArH).
3-羟基-3-正丙基苯并[c]硒吩酮的合成
在干燥的三颈瓶中放入0.55g(22.8mmol)镁粉和5mL无水乙醚,加入少量溴代正丙烷以引发反应,然后继续加溴代正丙烷直至镁粉完全消失,溴代正丙烷的加入总量为2.75g(22.8mmol),加热回流1h,冷却待用。将该格氏试剂中逐滴加入50mL含2.4g(11.4mmol)苯并[c]噻吩-1,3-二酮的无水乙醚溶液中,滴毕,加热回流1h,冷却室温搅拌8h。向此混合物中小心加入50mL饱和氯化铵溶液淬灭反应,然后加入5%稀盐酸调pH 2-3,分出有机相,水相再用乙醚萃取三次,合并有机相,无水硫酸钠干燥后蒸去溶剂。快速柱层析(石油醚/乙酸乙酯:20/1,V/V)得到黄色油状标题化合物1.05g,收率36%。ESI-MS:m/z 255[M-H]-;1H NMR(CDCl3,300Hz,δ):0.99(t,3H,CH3,J=7.5Hz),1.43-1.61(m,2H,CH2),2.10-2.19(m,1H,CH),2.39-2.50(m,1H,CH),7.45(m,1H,ArH),7.52-7.65(m,2H,ArH),7.79(d,1H,ArH,J=7.5Hz).
3-正丙基苯并[c]硒吩酮的合成
将0.52g(2.0mmol)3-羟基-3-正丙基苯并[c]噻吩酮溶于5mL冰醋酸中,0.5mL加入57%氢碘酸溶液,升温至125℃搅拌0.5-1h。冷却后向反应混合液中加入15mL 5%亚硫酸氢钠溶液,水层用乙醚萃取三次,合并有机相,无水硫酸钠干燥后蒸去溶剂。快速柱层析(石油醚/乙酸乙酯:50/1,V/V)得到淡黄色油状标题化合物0.42g,收率86%。ESI-MS m/z:241[M+H]+;263[M+Na]+;1H NMR(CDCl3,300Hz,δ):1.01(t,3H,CH3,J=7.5Hz),1.40-1.69(m,2H,CH2),1.83-1.96(m,1H,CH),2.37-2.49(m,1H,CH),5.11(m,1H,SeCH),7.39(t,1H,ArH,J=7.2Hz),7.52-7.63(m,2H,ArH),7.76(d,1H,ArH,J=7.8Hz).
实施例10
3-羟基-3-正丁基苯并[c]硒吩酮的合成
按实施例9的类似方法,使用2.55g(12.1mmol)苯并[c]硒吩-1,3-二酮、3.32g(24.2mmol)溴代正丁烷和0.58g(24.2mmol)镁粉进行反应,得到黄色油状标题化合物1.33g,收率41%。ESI-MS:m/z 269[M-H]-;1H NMR(CDCl3,300Hz,δ):1.06(t,3H,CH3,J=7.1Hz),1.41-1.61(m,2H,2×CH2),2.10-2.18(m,1H,CH),2.39-2.50(m,1H,CH),7.35(m,1H,ArH),7.51-7.65(m,2H,ArH),7.80(d,1H,ArH,J=7.5Hz).
3-正丁基苯并[c]硒吩酮的合成
按实施例9的类似方法,使用0.42g(1.6mmol)3-羟基-3-正丁基苯并[c]硒吩酮与0.5mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物0.30g,收率77%。ESI-MS m/z:277[M+Na]+;1H NMR(CDCl3,300Hz,δ):0.99(t,3H,CH3,J=7.2Hz),1.35-1.56(m,4H,2×CH2),1.74-1.87(m,1H,CH),2.26-2.35(m,1H,CH),5.10(m,1H,SeCH),7.45(m,1H,ArH),7.53(d,1H,ArH,J=7.5Hz),7.65(m,1H,ArH),7.79(d,1H,ArH,J=7.5Hz).
实施例11
3-羟基-3-正戊基苯并[c]硒吩酮的合成
按实施例9的类似方法,使用1.54g(7.3mmol)苯并[c]硒吩-1,3-二酮、2.20g(14.6mmol)溴代正戊烷和0.35g(14.6mmol)镁粉进行反应,得到黄色油状标题化合物0.71g,收率34%。ESI-MS:m/z 283[M-H]-;1H NMR(CDCl3,300Hz,δ):0.98(t,3H,CH3,J=6.9Hz),1.32-1.67(m,6H,3×CH2),2.16-2.23(m,1H,CH),2.40-2.52(m,1H,CH),7.45(m,1H,ArH),7.52-7.68(m,2H,ArH),7.80(d,1H,ArH,J=7.8Hz).
3-正戊基苯并[c]硒吩酮的合成
按实施例9的类似方法,使用0.45g(1.6mmol)3-羟基-3-正戊基苯并[c]硒吩酮与0.4mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物0.32g,收率75%。ESI-MS:m/z 269[M+H]+;291[M+N]+;1H NMR(CDCl3,300Hz,δ):0.90(t,3H,CH3,J=6.6Hz),1.34-1.60(m,6H,3×CH2),1.84-1.96(m,1H,CH),2.40-2.48(m,1H,CH),5.11(m,1H,SeCH),7.39(m,1H,ArH),7.52-7.63(m,2H,ArH),7.76(d,1H,ArH,J=7.5Hz).
实施例12
3-羟基-3-异戊基苯并[c]硒吩酮的合成
按实施例9的类似方法,使用3.25g(15.4mmol)苯并[c]硒吩-1,3-二酮、4.65g(30.8mmol)溴代异戊烷和0.74g(30.8mmol)镁粉进行反应,得到黄色油状标题化合物1.79g,收率41%。ESI-MS:m/z 283[M-H]-;1H NMR(CDCl3,300Hz,δ):0.90(m,6H,2×CH3),1.20-1.72(m,3H,CH2,CH),2.17-2.22(m,1H,CH),2.39-2.53(m,1H,CH),7.47(m,1H,ArH),7.53-7.63(m,2H,ArH),7.78(d,1H,ArH,J=7.5Hz).
3-异戊基苯并[c]硒吩酮的合成
按实施例9的类似方法,使用0.34g(1.1mmol)3-羟基-3-异戊基苯并[c]硒吩酮与3mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物0.24g,收率79%。ESI-MS:m/z 269[M+H]+;291[M+Na]+;1H NMR(CDCl3,300Hz,δ):0.89(m,6H,2×CH3),1.25-1.70(m,3H,CH2,CH),1.84-1.97(m,1H,CH),2.41-2.53(m,1H,CH),5.11(m,1H,SeCH),7.39(m,1H,ArH),7.53-7.63(m,2H,ArH),7.77(d,1H,ArH,J=7.5Hz).
实施例13
3-羟基-3-正己基苯并[c]硒吩酮的合成
按实施例9的类似方法,使用3.75g(1.78mmol)苯并[c]硒吩-1,3-二酮、5.86g(3.55mmol)溴代正己烷和0.85g(3.55mmol)镁粉进行反应,得到黄色油状标题化合物1.85g,收率35%,m.p.65-67℃。ESI-MS:m/z 297[M-H]-;1H NMR(CDCl3,300Hz,δ):0.89(t,3H,CH3,J=7.1Hz),1.32-1.57(m,8H,4×CH2),2.19-2.24(m,1H,CH),2.40-2.52(m,1H,CH),7.46(m,1H,ArH),7.52-7.63(m,2H,ArH),7.80(d,1H,ArH,J=7.8Hz).
3-正己基苯并[c]硒吩酮的合成
按实施例9的类似方法,使用0.45g(1.5mmol)3-羟基-3-正己基苯并[c]硒吩酮与6mL57%氢碘酸溶液反应,得到淡黄色油状标题化合物0.29g,收率69%。ESI-MS:m/z 283[M+H]+;305[M+Na]+;1H NMR(CDCl3,300Hz,δ):0.87(t,3H,CH3,J=7.2Hz),1.25-1.65(m,8H,4×CH2),1.84-1.96(m,1H,CH),2.44-2.48(m,1H,CH),5.10(m,1H,SeCH),7.39(m,1H,ArH),7.52-7.62(m,2H,ArH),7.76(d,1H,ArH,J=7.8Hz).
一种3-取代苯并[c]呋喃酮的硫代、硒代同系物、其制备方法及医药用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0